Animal models of inflammatory bowel disease: novel experiments for revealing pathogenesis of colitis, fibrosis, and colitis-associated colon cancer
Article information

Abstract
Inflammatory bowel disease (IBD), comprising Crohn’s disease and ulcerative colitis, is a lifelong disease that manifests with chronic intestinal inflammation, sequential fibrosis, and an increased risk of colitis-associated colon cancer (CAC). The combined effects of genetic, immunological, environmental, and microbial factors render it difficult to determine the specific mechanism underlying the induction and perpetuation of IBD. Various animal models of IBD have contributed enormously to the understanding of IBD pathogenesis in terms of genomics, transcriptomics, proteomics, microbiome, and drug development of novel therapeutics. Although comprehensive research on IBD has been enabled by advanced technologies, such as genetically engineered models, there is a great need to develop relevant in vivo models of colitis and fibrosis. Here, we review 4 categories of animal models of acute and chronic intestinal inflammation, fibrosis, and CAC: chemically induced, genetically engineered, T cell transfer, and spontaneous gene mutation models.
INTRODUCTION
Inflammatory bowel diseases (IBDs), including Crohn’s disease (CD) and ulcerative colitis (UC), are traditionally regarded as diseases in Westernized nations affecting over 1.5 and 2 million people in the United States of America and Europe, respectively [1,2]. However, at the turn of the 21st century, the epidemiology of IBD is changing worldwide, due to rapid socio-economic development [3]. Epidemiological reports suggest that the incidence of IBD is rising rapidly in South America, Asia, and Africa [4]. It has become a global economic concern in terms of resource utilization and healthcare costs [5]. IBDs, symptomatically characterized by chronic diarrhea, abdominal pain, gastrointestinal bleeding, and weight loss, are idiopathic chronic and relapsing immunoinflammatory disorders of the gastrointestinal tract. Patients with IBDs show various disease courses owing to chronic inflammation, resulting in fibrosis and colitis-associated colon cancer (CAC).
The etiology of IBD remains unclear, although numerous studies have attempted to reveal the factors underlying the pathogenesis of IBDs over the last three decades when experimental models of intestinal inflammation that resemble IBD have become popular [6,7]. However, the accumulation of epidemiologic, genetic, and clinical studies in patients with IBD, as well as animal IBD models, suggests that a complex combination of causative factors such as genetic, environmental, microbial, and immunologic factors are related to the initiation and perpetuation of intestinal inflammation [8,9]. Owing to multifactorial etiology and heterogeneous phenotypic symptomatology, there is no single translatable experimental animal model that entirely represents human IBD pathophysiology and related complications, such as fibrosis and CAC. In addition, there is a need to develop relevant in vivo models of IBD to translate the preclinical effects of novel drugs into clinical treatment options.
Recently, numerous murine models of colitis have been developed and implemented. Each murine model has its strength in unraveling the pathogenesis of colonic inflammation, fibrosis, or CAC. However, these models have limitations, such as a self-limiting nature and marked variability in colitis development. Therefore, it is imperative to select an appropriate animal model and standardize experimental design. In this review, we provide an update on animal models of IBDs, highlighting each method in terms of acute/chronic colitis, intestinal fibrosis, and CAC research.
CHEMICALLY-INDUCED MODELS
In 1957, an experimental colitis model was first devised using crystalline egg albumin, which sensitizes rabbits with dilute formalin [10]. Since then, many other types of chemically-induced colitis models have been developed, mainly in rats, mimicking several key immunological and gut histopathological features of human IBDs [11]. These models could be divided into those that disrupt the gut mucosal/epithelial barrier and/or that involve hapten-induced hypersensitivity reactions. In addition to the types of models, each chemically-induced model can be used to investigate intestinal fibrosis and inflammation-associated carcinogenesis, as summarized in Table 1.
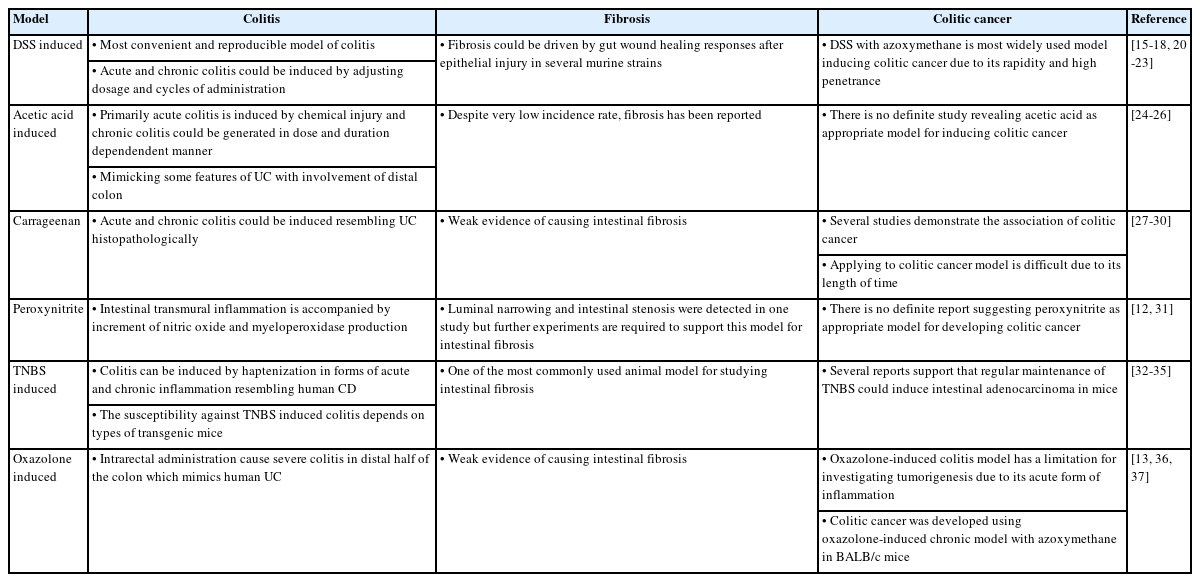
Synopsis of Chemically-Induced Models in Aspects of Colitis, Fibrosis and Colitis-Associated Colon Cancer
Advantages: The primary advantage of these models is the ease of induction, reproducibility, rapidity, and capability of using healthy wild-type mice as well as genetically engineered mice. Thus, they have been widely adopted to identify the key factors underlying intestinal inflammation and cytokines dysregulation. Moreover, compared with genetically engineered models, these models can avoid developmental defects in laboratory animals by genetic modification or high variability in penetrance and severity of colitis between animals.
Limitations: Inducing self-limiting inflammation rather than a chronic phase remains a limitation that cannot fully describe the complex pathogenesis of IBDs in humans. Several reports attempted to overcome this limitation of the acute inflammatory phase as modification of dosages and cycles allows eventual relapsing and chronic forms of intestinal inflammation and fibrosis [12,13]. With the standardized experimental design, careful selection among diverse chemically-induced models are suggested to ensure translational value.
1. Disruption of Mucosal/Epithelial Barrier
1) DSS-Induced Murine Models for Acute and Chronic Colitis
Dextran sulfate sodium (DSS) is a water-soluble chemical. The colitogenic potential depends on its molecular mass, which ranges from 5 to 1,400 kDa [14]. Of note, 40 to 50 kDa is known as the appropriate molecular weight for colitis induction. The DSS solution is typically prepared on the administration day with a magnetic stirrer to ensure complete dissolution, with the DSS concentration varying between 3% and 5%. Before administration of the DSS solution, the mice are weighed. After administering the DSS solution via drinking water, researchers should monitor the body weight, stool consistency, hematochezia, and activity of the mice. The duration of DSS administration can be determined according to body weight and disease activity index (DAI) assessed by weight loss, stool consistency, and hematochezia for colitis.
Oral administration of DSS causes chemical damage to the epithelial monolayer of intestinal tissue, which affects the immune response and causes mucosal barrier dysfunction. The DSS-induced murine model exhibits human UC-like histopathology mainly in distal colon segments, such as colon and cecum shortening, erosions, ulcers, crypt loss, and neutrophil infiltration, as well as weight loss, occult blood in feces, and bloody diarrhea. Importantly, T and B cells are minimally required for the induction of intestinal inflammation in this model, as shown in a study with strains characterized by the absence of adaptive immunity, such as severe combined immunodeficiency and Rag−/− mice [15,16]. Although the mechanism differs substantially from that of colitis in human IBD, it is worth analyzing the innate immune system in the development of colitis and regeneration of injured gut epithelium.
Owing to its self-limiting properties, this model is appropriate for revealing key cellular events and cytokines in acute inflammatory settings. However, a chronic DSS colitis model has been successfully established. Several studies have revealed that oral administration of DSS via the drinking water of BALB/c mice and Swiss–Webster mice causes chronic colitis after multiple cycles of DSS [17,18]. In addition, dysregulation of T helper 1 (Th1)/T helper 2 (Th2) balance has been shown over time in chronic DSS colitis [19].
2) A DSS-Induced Murine Model for Fibrosis
The DSS-induced fibrosis model could be driven by gut wound-healing responses developed immediately after an acute epithelial injury in the study with FVB-N wild-type mice and C57BL6 procollagen α1(I)-GFP reporter mice [20]. Furthermore, inducing chronic inflammation with several cycles of DSS reportedly triggers fibrosis [21]. In certain strains, a substantial amount of fibrosis has been observed. However, the relationship between the DSS-induced model and fibrosis in human CD remains controversial. There is insufficient evidence to suggest that chemical damage results in fibrotic complications in patients diagnosed with CD.
3) DSS with Chemical Carcinogens
Compared to the administration of DSS alone, colorectal cancer following colitis can be rapidly initiated by adding colon carcinogens, such as heterocyclic amines, aromatic amines, alkylnitrosamide compounds, 1,2-dimethylhydrazine, and azoxymethane (AOM) [22]. Each compound has its own advantages and limitations according to its effects on histogenesis, cell proliferation kinetics, genetics, and environmental traits. As one of the most commonly used colonic carcinogen, AOM resembles human colorectal cancer in terms of histopathological and proteomic features, and frequent mutations in K-Ras and β-catenin [38-40]. Resulting in the rapid development of intestinal tumors per mouse within 10 weeks, a combination of DSS with AOM is practically suitable for the colitic cancer model in IBDs [23]. Beyond dose dependency, sensitivities to the DSS with AOM administration reportedly vary in different murine strains. In a study of BALB/c, C3H/HeN, C57BL/6N, and DBA/2N strains, BALB/c mice showed the highest tumor frequency (100%) after a single AOM injection (10 mg/kg) and 1% DSS for 4 days [41]. In another investigation with weekly injections of AOM (15 mg/kg), BDIX/OrlIco mice showed a higher incidence (75%–100%) of tumor development compared to F344/NHsd and WAG/Rij strains [42].
4) Other Types of Mucosal-Disrupting Agents for Inflammation and CAC
In addition to DSS, many other chemicals, such as acetic acid, ethanol, carrageenan, and peroxynitrite, have been introduced to IBD animal models with improved methodology. In 1978, acetic acid was reported as a chemical substance that induces colonic epithelial injury followed by inflammatory cell infiltration [24,25]. Acute colitis was induced by chemical injury instilled via the rectum. Notably, although it has a low incidence rate, chronic inflammation and fibrosis can also be generated in a dose- and time-dependent manner [26]. Ethanol has been widely used as a breaking mucosal barrier that enables the haptenizing agent to induce a hypersensitivity immune response. Carrageenan is a highly sulfated hydrophilic colloid that induces acute and chronic colitis, characterized by mucosal ulceration, gland distortion, and crypt abscess resembling human UC. Activation of B-cell lymphoma 10 and nuclear factor kappa B signal transduction pathways and the involvement of Toll-like receptor 4, interleukin (IL)-6, and IL-8 have been shown in this colitis model [27,28]. In addition, carrageenan could reduce certain anti-inflammatory bacteria in a murine model [43]. Several reports have demonstrated an association between degraded carrageenan in the diet and colonic tumor occurrence. However, these studies took a long time to generate colon tumors, rendering this model difficult to utilize in experimental colitis models [29,30]. Promoting lipid peroxidation and sulfhydryl oxidation, peroxynitrite experimentally causes transmural inflammation and fibrosis. In a murine model, luminal narrowing and intestinal stenosis were detected on days 7 and 21, respectively, after enteral peroxynitrite administration [31].
2. Hapten-Induced Hypersensitivity Reaction
1) TNBS/DNBS in Ethanol for Inflammation, Fibrosis, and CAC
2,4,6-Trinitrobenzene sulfonic acid (TNBS) and dinitrobenzene sulfonic acid (DNBS) are contact-sensitizing agents that cause T cell-mediated transmural colitis that resembles CD in humans following the intrarectal administration of ethanol. Once administered, ethanol primarily damages the intestinal mucosa and allows TNBS and DNBS to penetrate the bowel wall. Subsequently, the haptenization of colonic or microbiota-derived proteins causes the generation of trinitrophenyl-specific B cells and Thl -type responses. A single dose of rectal administration results in an acute inflammatory reaction by activating Th1-related cytokines. It has been reported that infiltration of inflammatory cells in colon tissue occurs within 2 hours. In addition, the delayed-type hypersensitivity reaction can be driven by the second dose of TNBS approximately 6 days post-induction [32]. The concentration of TNBS varies among reports, and the DAI and body weight should be measured daily. Generally, mice are euthanized after 3 days of rectal enema based on weight and DAI.
Weekly rectal enema of TNBS, which takes approximately 45 to 49 days, induces chronic colitis characterized by increased IL-17 and IL-23 levels [33]. The susceptibility toward TNBS-induced colitis significantly depends on the type of transgenic mice. It has been reported that SJL/J and BALB/c mice are susceptible to TNBS whereas C57BL/6 and 10 mice are resistant [34]. Owing to the susceptibility, SJL/J strain has mainly been used in the TNBS colitis model. It is also known that adenocarcinoma can be induced by TNBS. CAC developed in the group administered TNBS twice a week after induction. Dysplasia and carcinoma were detected at 5 weeks [35].
The TNBS-induced model is used most commonly for studying intestinal fibrosis during the early or late course of the disease. Repetition and escalation of TNBS dosage over 6 weeks could induce fibrosis in the chronic colitis model. Several studies have shown an increased amount of collagen in colon tissue and architectural intestinal fibrosis by Masson’s trichrome tissue staining in BALB/c mice. Transforming growth factor (TGF)-β1 is regarded as a crucial factor that powerfully drives fibrosis in the gut as well as in essentially all other organs [44]. It has been revealed that Th2 cytokines and IL-13-induced TGF-β1 productions are predominantly increased in the late stage of inflammation, whereas IL-12p70 and interferon-γ concentrations increase in the early stage [45]. In addition, TNBS-induced fibrogenesis is also nuclear factor kappa B activation-dependent [46]. The advantage of the TNBS model for studying intestinal fibrosis is the ease of the experimental execution and the presence of certain features of CD. However, this model should be chosen carefully, considering the location of inflammation and fibrosis that might be restricted in the distal colon due to rectal administration of enema of TNBS and the potential for results variability among murine strains used.
2) Oxazolone in Ethanol
Oxazolone is a classical haptenating agent with a colitogenic potential. It has been used to induce acute and chronic colitis in murine models. Treatment with oxazolone on days 1 and 8 is the usual protocol for the acute colitis model, while administration on days 1, 6, 20, and 34 resulted in chronic colitis. Unlike the TNBS/DNBS-induced colitis model, intrarectal administration of ethanol causes severe colitis in the distal half of the colon, mimicking human UC rather than CD. It has been known that the C57BL/6 strain is resistant to oxazolone-induced colitis while SJL/J and C57BL/10 mice are highly susceptible. Histologically, it is characterized by damage to the mucosa and submucosa with mixed cell (neutrophil, macrophage, and lymphocyte) infiltration and has the feature of IL-4-driven Th2-type colitis rather than IL-12-driven colitis. Through lymphocyte activation and IL-9 production, the IL-4R signaling pathway plays a key role in this model. In addition, the administration of anti-IL-4 agents prevents oxazolone-induced colitis. Compared to TNBS, oxazolone also induces a TGF-β response, limiting the extent and duration of colitis [36]. This model has a limitation in investigating tumorigenesis due to its acute nature. However, CAC can be induced in a chronic model using BALB/c mice [37].
GENETICALLY ENGINEERED MODELS
In 1993, spontaneous colitis in knockout (KO) mice was discovered, including IL-2 KO, IL-10 KO, and T cell receptor (TCR) KO mice [47]. Contemporary gene mutation models that induce colitis and/or ileitis spontaneously include over 60 different kinds of KO mouse models, as shown in Table 2. Several types of genetically engineered models can be distinguished by these precise methods and the extent of their influence. The conventional model of KO mice is modified to lack a specific gene in all cell types, including all other organs, such as the IL-10 KO model [48]. To overcome this limitation, technology that allows modification of particular genotypes only in cells of the targeted organ was invented in the 1990s and the 2000s, referred to as cell-specific or conditional KO models. Using the Cre-lox recombination system, it is possible to make a deletion of genes and its inversion or complete inactivation. Furthermore, technologies such as reversibly controlling expression, overexpressing by introducing genes of interest in all cells or specific types of cells, and interfering with overexpressed non-functional proteins with a target factor, have been developed. In addition to selecting methods for genetically engineered models, it is crucial to decide which genes to target. Genotypic factors that influence epithelial barrier function, T and/or B-cell regulation, and signal transduction for inflammation should be selected appropriately for each IBD model.

Knockout Mouse Models for Inflammatory Bowel Disease
Advantages: These novel technologies allow researchers to analyze the genetic influence on the development of colitis that mimics IBDs. Overexpression and deficiency of specific genes in animal models could generate knowledge of particular functions and mechanisms of interesting substances in intestinal inflammation, fibrosis, and CAC. Moreover, examining pathogenic or regulatory factors is also possible during the development of chronic colitis in genetically confined murine models.
Limitations: Genetically engineered models cannot be a complete surrogate for human IBD, which has diverse risk factors beyond the genetic background. In addition, considerable variability in the development of intestinal inflammation can exist between facilities because of different microbial environments. There is also the limitation of prolonged disease development time compared with other animal models.
1. Genetically Engineered Models of Chronic Colitis
IL-10 is a regulatory cytokine that plays key roles in immunosuppression and inflammation. Deletion of the IL-10 gene causes genetically engineered mice to spontaneously develop intestinal inflammation after 3 months of age [49]. In addition, nonsteroidal anti-inflammatory drugs such as piroxicam and sulindac have been recently used to accelerate and synchronize the onset of colitis [50]. Spontaneous and unremitting inflammation is driven by a Th1 T cell response that causes infiltration of lymphocytes, macrophages, and neutrophils in the colon. It has been revealed that the enteric microbiome plays a crucial role in immune system activation in IL-10 KO mice. In several reports, mild colitis occurred under specific pathogen-free conditions, and colitis was not induced under germ-free conditions [51,52].
2. Genetically Engineered Models of Fibrosis
It has been reported that intestinal fibrosis could be induced in a model of IL-10 deficiency and overexpression of TGF-β1 and monocyte chemoattractant protein 1 (MCP-1). In the late phase of colitis, the extracellular matrix accumulates in the damaged intestine with profibrotic cytokine levels such as IL-13 [21,53]. TGF-β1 is a key cytokine in the development of colon fibrosis in IBD. Bioactive TGF-β1 can be transferred by an adenoviral vector, which leads to the development of submucosal fibrosis. Genetic ablation of TGF-β1 results in a fatal outcome 5 weeks after birth, suggesting that the TGF-β1-suppressing method for anti-fibrogenesis seems to be dangerous. A murine model of TGF-β1 overexpression is valuable for studying intestinal fibrosis. MCP-1 is a well-known cytokine that causes fibrosis in other organs. In the colon targeting murine model, intramural delivery of the adenoviral vector encoding murine MCP-1 induced collagen accumulation and transmural inflammatory cell infiltration between days 3 and 21 [54].
3. Genetically Engineered Models of CAC
Several experiments have suggested that KO and transgenic murine models can unveil the concepts and details of the pathogenesis of chronic inflammation toward colon cancer. Transfection with a dominant negative N-cadherin mutant causes porous intestinal epithelial cells, resulting in chronic colitis and neoplasia. Sadlack et al. [55] found that IL-10/β2-microglobulin double-KO mice develop intestinal carcinoma after modest colitis with a long life span. In addition, deficiencies in TCR-related genes can cause spontaneous chronic colitis, dysplasia, and adenocarcinoma. It has been reported that spontaneous adenocarcinoma of the large intestine develops in the TCR-β chain and transformation-related protein 53 double-KO mouse strain [56,57].
T CELL TRANSFER MODEL
Numerous experimental studies have suggested that T cell-dependent models of IBD may be more appropriate for human chronic colitis than self-limiting models of erosive colitis because of the critical role of the dysregulated immune response. In 1990, a novel method using a T cell transfer system was introduced, contributing tremendous efforts to accumulate knowledge of regulatory T cells [58,59]. While the onset and severity of colitis are relatively variable in genetically engineered models such as IL-10 KO mouse model, the T cell transfer model of chronic colitis could meet the need to precisely synchronize the onset and severity of inflammation. Small and large intestinal inflammation is induced after 5 to 10 weeks by transferring naïve CD4+ T cells (CD4+ CD45RBhigh T cells) into syngeneic recipients that lack both T and B lymphocytes, such as RAG1 KO, RAG2 KO, and severe combined immunodeficiency mice [60]. In contrast, transferring mature CD4+ T cells (CD4+ CD45RBlow T cells) with or without naïve CD4+ T cells into recipient mice could not induce colitis [61,62]. The T cell transfer model of colitis is caused by Th1 responses related to interferon-γ and tumor necrosis factor α presenting as transmural inflammation, crypt abscesses, epithelial cell hyperplasia, and erosions. In addition to pathogenic CD4+ T cells, CD3εTg26- and hsp60-specific CD8 T cells have also been reported to trigger colitis via adoptive transfer [63,64].
Advantages: Adoptive T cell transfer animal model closely reflects the pathophysiology of IBD, especially T cell migration to the intestine. These models are capable of revealing the earliest immunological mechanism and role of regulatory T cells in the induction and perpetuation phases of colitis. Furthermore, the T cell transfer system has been used to demonstrate the effects of pharmaceutical agents on induced fibrosis [65].
Limitations: These models are relatively difficult to establish because of their complex experimental process and time-consuming nature. Owing to the use of immunodeficient mice, integrated knowledge of the generation of colitis around complex factors could not be obtained.
SPONTANEOUS GENE MUTATION MODELS
Without any genetically modifying intervention or exogenous manipulation, few mouse strains exhibit spontaneous colitis resembling human IBDs. The SAMP1/YitFc strain develops spontaneous colitis at 10 to 20 weeks of age [66,67]. Showing 100% penetrance in all animals, mice aged 30 to 40 weeks typically develop persistent severe ileitis. Characterized by affecting the terminal ileum and segmental transmural inflammation, this model resembles CD. Moreover, perianal fistula and intestinal stricture formation were observed in some mice in this model. Interestingly, it has been revealed that the SAMP1/YitFc strain cannot generate colitis in germ-free conditions [68]. Due to the low breeding rate and commercial unavailability of SAMP1/YitFc mice, significant limitations persist for researchers to perform large-scale experiments. In addition to the SAMP1/YitFc mouse model of colitis, the C3H/HeJBir strain also develops spontaneous colitis owing to the absence of Toll-like receptor 4. Increased reaction of CD4+ T cells and B cells toward bacterial antigens results in transmural colitis at 3 to 4 weeks of age [69,70].
BACTERIA-INDUCED MODELS
Considering the important role of the microbiome in the initiation and perpetuation of chronic colitis in IL-10 KO murine models, several studies have introduced animal models infected with specific or complex microbiomes into novel experiments. Injection of fecal suspension or selected bacteria, such as Lactobacillus ssp., Enterobacter aerogenes, Streptococcus viridans, Clostridium ramosum, Bacteroides fragilis, and Bacteroides uniformis into the colonic wall of rats was performed. Chronic colitis and intestinal fibrosis occurred in all the groups, with increased amounts of collagen and high levels of TGF-β1. Moreover, these models showed an increased proportion of bowel strictures in infected rats [71]. Oral administration of Salmonella enterica serovar Typhimurium after 24 hours of streptomycin treatment in mice resulted in edema, ulceration, and even transmural intestinal inflammation. Furthermore, fibrosis was observed after 7 days of infection with deposition of type I collagen [72]. Those experiments provided important insight and potential for investigation of the bacterial contribution to IBD in aspects of chronic colitis and fibrosis.
SELECTION OF ANIMAL MODELS AND STUDY DESIGN
Currently, several animal models of disease are available for IBD research. It is quintessential to select the relevant mouse model because each murine model has a different type of intestinal inflammation in the small and large bowels. However, there are no standardized methods for selecting a single appropriate model to obtain a reasonable answer to the scientific question. Instead, some strategies can be suggested to determine a suitable model. It is inevitable to be concerned about the costs and experimental facilities before making decisions regarding particular animal models. For example, novel genetic engineering techniques such as the Cre-lox recombination system are comparably more expensive than experiments using chemicals. Moreover, preclinical studies utilizing germ-free conditions require certified facilities and skilled researchers. In affordable and available circumstances, it is recommended to consider each characteristic of the individual model in terms of anatomy, evoked nature, dominant pathological response, and the ultimate purpose of the experiment and hypothesis being tested. As CD and UC can affect different anatomical regions, such as the small intestine, colon, and rectum, regional differences should be considered in selecting models. Acute colitis induced by chemicals such as acetic acid and oxazolone administered via the rectum results in inflammation of the distal colon which is not suitable for studies on CD. In addition, the acute or chronic nature of intestinal inflammation and prevalent pathological responses, such as innate or adaptive immune responses, could be crucial factors to be considered. Finally, a model that fits well with the ultimate purpose must be carefully chosen, including studies of transmural inflammation, fibrosis, and colitis-associated cancer.
CONCLUSION
With the development of experimental methods utilizing animal models, tremendous knowledge of IBD has been gained in terms of pathogenesis, treatment, and prognosis. Nevertheless, it remains a challenge to translate preclinical results into new drug development. Although intestinal fibrosis and malignancy are the main complications of IBD, pathogenic factors have not been fully revealed or used as therapeutic targets. Currently, the only available treatment for fibrostenotic and malignant complications is surgery. To overcome these issues, preclinical studies should be performed using relevant animal models with appropriate designs. In addition, numerous studies can be conducted, including genomics, transcriptomics, proteomics, and the microbiome based on animal models. Integration with emerging technologies such as organoids and humanized mouse models can help overcome previous limitations of preclinical studies. In order to control the inherent constraints of preclinical studies, it is also recommended to design experimental studies in the combination of animal research and human studies. These will lead to the development of new therapeutic agents for fibrosis and prevention of CAC, eventually helping improve clinical outcomes in patients with IBD.
Notes
Funding Source
Koh SJ was supported by a National Research Foundation of Korea grant funded by the Korea government (MSIT; No. 2019 R1C1C1002243) and the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2016R1D1A1B03931961 and NRF-2020R1A1F10666419, NFR-2020R1F1A1066491).
Conflict of Interest
Radi ZA and Habtezion A are employees of Pfizer Inc. No potential conflict of interest relevant to this article was reported.
Data Availability Statement
Not applicable.
Author Contribution
Conceptualization: Koh SJ. Data curation: Lee CH. Investigation: Lee CH. Resources: Lee CH, Koh SJ. Supervision: Radi ZA, Habtezion A. Visualization: Lee CH. Writing - original draft: Lee CH, Koh SJ. Writing - review & editing: Lee CH, Koh SJ, Radi ZA, Habtezion A. Approval of final manuscript: all authors.