Delineating inflammatory bowel disease through transcriptomic studies: current review of progress and evidence
Article information
Abstract
Inflammatory bowel disease (IBD), which comprises of Crohn's disease and ulcerative colitis, is an idiopathic relapsing and remitting disease in which the interplay of different environment, microbial, immunological and genetic factors that attribute to the progression of the disease. Numerous studies have been conducted in multiple aspects including clinical, endoscopy and histopathology for the diagnostics and treatment of IBD. However, the molecular mechanism underlying the aetiology and pathogenesis of IBD is still poorly understood. This review tries to critically assess the scientific evidence at the transcriptomic level as it would help in the discovery of RNA molecules in tissues or serum between the healthy and diseased or different IBD subtypes. These molecular signatures could potentially serve as a reliable diagnostic or prognostic biomarker. Researchers have also embarked on the study of transcriptome to be utilized in targeted therapy. We focus on the evaluation and discussion related to the publications reporting the different approaches and techniques used in investigating the transcriptomic changes in IBD with the intention to offer new perspectives to the landscape of the disease.
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic, relapsing and remitting inflammatory gastrointestinal disorder comprises of 2 dominant sub-types which are CD and UC. CD affects any component of the gastrointestinal tract starting from the oral cavity to the anus with transmural inflammation.12 In contrast, UC affects the colon and characterized by the inflammation of the mucosa and submucosa layers of the rectum and colon accompanied by cyptitis.3 It is thought that several factors contributed to the development of the disease including environment, genetics, gut microbiota and immune response to intestinal microbiota.245 However, at this point of time, there is an insufficient research data to make a definitive conclusion on the causes of IBD.
The incidence of CD has increased steadily in the North America and Europe ranged from 0 to 20.2 cases per 100,000 population while the incidence of UC ranged from 0 to 24.3 per 100,000 population.6 However, in recent years, there has been an increasing amount of literatures reporting on the sharp rise in the incidence of IBD in developing countries especially in Asia.78910 The incidence of IBD in Asian countries ranged from 0.5 to 3 per 100,000 population with UC having a higher incidence as compared to CD, approximately 1 and 0.5 per 100,000 populations respectively.11 Recent researches have suggested that urbanizations including Western dietary pattern, hygiene and childhood immunological factors are associated with IBD in Asia.1213 Some other reports have identified that the changes in the gut microbiota and environmental factors are also linked to the causes of IBD.91415
However, apart from the aforementioned factors, a substantial number of studies had also reported on the involvement of genetic variations in the pathogenesis of IBD.1617 There has been a surge of interest for the past few years in studying transcriptomic dysregulations in IBD whereby the changes involving all the transcribed RNA transcripts from the genomes.31819 Transcriptomic studies are mainly dominated by arrays and sequencing technologies where vast information on the functions, expression levels and biological pathways of different transcripts are being analyzed with regards to IBD.18192021
WHAT IS TRANSCRIPTOME?
The term transcriptome is defined as a small proportion of the genetic codes that is transcribed into RNA molecules.22 It is estimated that less than 5% of the genome in human is transcribed into the RNA molecules.23 Interestingly, the proportion of transcribed RNA sequences that belongs to the nonprotein coding RNA groups appears to be in a greater percentage in a complex organism.24 Transcriptome is generated through the transcription process whereby DNA sequences are copied into complementary RNA strands by RNA polymerase. These complementary RNA strands, which consists of introns and exons, will undergo splicing to remove the introns in order to generate mature RNA transcripts that contain only the exons.25
Post-transcriptional RNA processing includes alternative splicing, RNA editing and various combinations of transcription initiation and termination sites are crucial processes in the cells as this could produce more than one variants of mRNA. In consequence, different protein products can be generated from a single transcribed gene.25 Therefore, investigations on the whole transcriptome would provide a better understanding on the complexity of the disease as compared to a single genomic study.
DIFFERENT TYPES OF RNAS AND THEIR ASSOCIATIONS WITH DISEASES
RNAs have been widely explored and have provided significant amount of information for a better understanding of IBD pathogenesis. RNAs are divided into different classes, including mRNA, transfer RNA (tRNA), ribosomal RNA (rRNA) and noncoding RNA (ncRNA). Examples include riboswitches, ribozymes, long noncoding RNA (lncRNA) and microRNA (miRNA).26 A diagrammatic representations of different classes of RNAs in eukaryotic cells involved in IBD is as shown in Fig. 1. Mature mRNA contains coding information for only one polypeptide chain which comprises of a cap, a coding region with exons and a tailing sequence that includes the poly(A) tail.27 The translation of mature mRNA into its specific protein is controlled by poly (A)-binding protein that binds to the mRNA's poly A tail.28 Based on these RNA characteristics, advanced techniques such as next-generation sequencing (NGS), transcriptomic array, TaqMan gene expression array and microarray were established and used in the high throughput data analysis of mRNA.3162930 As mRNAs contain information for proteins formation and different pathways regulation, an increasing amount of researches have been accomplished worldwide in exploring the transcripts sequences, functions, translation, expression level and their influences in different kind of diseases.313233 Examples of diseases where their pathogenesis had been studied in terms of the dysregulations of mRNAs include schizophrenia, cancer, autoimmunity, neurological disorders and diabetes.173435 In transcriptomic studies of IBD, most of the aberrant mRNAs were found to be involved in molecular functions associated with immune response, mucosal inflammation, nutrients absorption, epithelial damage, oncogenesis and cell proliferation.213637

A representation of different types of RNAs in eukaryotic cells. Different classes of matured RNAs transcribed from genomic DNA are involved in the regulation of different cellular processes and their dysregulations could lead to numerous diseases including IBD. rRNA, ribosomal RNA; tRNA, transfer RNA.
rRNA, which is a part of ribosome (a protein synthesizing organelle), plays an important role in translating the information in mRNA to protein in the cytoplasm in a process called translation. In human genome, there are approximately 300 to 400 copies of rRNA genes and they are organized in a repeated tandem arrays within nucleolar organizing regions located on the short arm of chromosome 13, 14, 15, 21 and 22.38 Variations in rRNA genes and its expression level has been reported to be associated with carcinogenesis, pathogenesis of schizophrenia and autism as well as in hearing loss.3940
tRNAs are the adaptor molecules for accurate mRNA translation for protein synthesis. The mature form of tRNAs consists of approximately 70 to 100 nucleotides long known as “clover” leaf secondary structure and folds into a common L-shaped architecture.41 These amino acid carriers decode the nucleotide sequence of mRNA into specific polypeptide sequences thus allowing the genetic code to be specifically translated for protein synthesis.42 Additional role of tRNAs have been reported in stress response, gene regulation and plasmid replication.4344 Similarly to rRNA study, there has been no report on the involvement of the tRNA on the pathogenesis of IBD.
ncRNAs are described as RNA molecules that are non-templates for protein synthesis and are expressed as small or lncRNAs.45 lncRNAs are generally defined as ncRNAs that are greater than 200 nucleotides in length.46 A great deal of previous researches into lncRNAs have focused on the functions of this molecule on numerous cellular processes including cell cycle regulation, stem cell pluripotency, retrotransposon silencing, meiotic entry and telomere length.4748 Thus, it is reported that lncRNAs are able to regulate gene expressions including the neighboring protein-coding genes.495051 In IBD, overexpression of lncRNAs were reported to be related to immune response, pro-inflammatory cytokines activity and major histocompatibility protein complex.3
miRNAs belong to a family of small ncRNAs (20–24 bps) and play important roles in human as they are involved in the post-transcriptional stage of gene expression.5253 miRNA represses gene function by binding at the 3′ untranslated region (UTR) of the targeted mRNA and subsequently leads to the prevention of mRNA translation or degradation (Fig. 2). The roles of miRNAs have been described in cellular processes including proliferation, apoptosis, development and cell fate programming.545556 It has been demonstrated that altered miRNAs expressions are observed in a number of diseases such as in cancer and cardiovascular disorders, myocardial infarction, heart failure, acute coronary syndrome and IBD.575859 Numerous miRNAs were found to be differentially expressed in IBD and were hypothesized to act as critical mediators in the IBD pathogenesis by regulating proteins in vital regulatory pathways such as in the expression of inflammatory cytokines.60
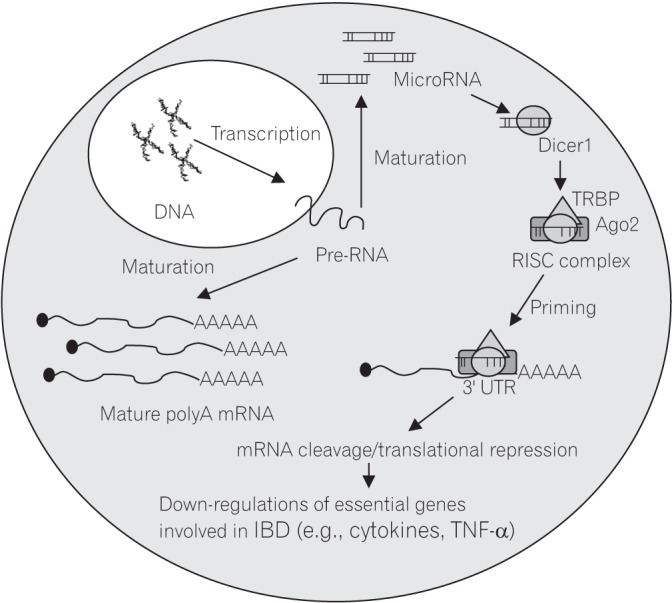
Mechanism of action of microRNA in repressing the essential genes involved in the regulation of inflammatory processes in epithelial cells through the binding at the 3′ UTR of the genes. Dicer1, RNAse endonuclease; RISC complex, RNA-induced silencing complex; TRBP, protein with 3 double-standard RNA-binding domains; Ago2, Argonaute 2; 3′ UTR, 3′ untranslated regions; TNF-α, tumour necrosis factor α.
TRANSCRIPTOMIC DYSREGULATION STUDIES IN IBD
Various experimental designs on the study of transcriptomic dysregulations have been tailored and carried out in IBD patients whereby novel findings, objectives or correlations between the studied cohorts could be discovered in terms of the changes in RNAs expression. All the different approaches and studies discussed here are summarized in Table 1.31821376162 One of the common achievements includes the genome-wide microarray study identified different types of aberrant RNAs and the deregulation of molecular mechanisms directly involved in IBD, including the complex array of pathways from inflammatory responses to the deregulations of growth factors. For example, a comprehensive high-throughput mRNA expression study by Costello et al.37 had reported that a total of 500 and 272 transcripts were differentially regulated in CD and UC patients respectively. The gene dysregulation study was conducted using genome-wide cDNA microarrays with biopsies taken from normal (n=11), CD (n=10) and UC (n=10) patients followed by verification of the gene expression of interesting hits with real-time quantitative PCR (qPCR) in additional samples of 100 individuals. From the results, there were 122 genes found to be dysregulated in both conditions and on average ~40% of these identified differentially regulated genes are novel and unannotated.37 These differentially upregulated genes were found to be involved in the functional category of “immune and inflammatory response,” “oncogenesis,” “cell proliferation and growth” and “structure and permeability” whereby a transcribed gene sequence with strong similarity to protein pir:I38067, nitric-oxide synthase, and the Ig heavy constant γ 1 (IGHG1) gene is reported to be upregulated by more than 10-fold in both IBD cases against normal patients. While for another aberrant gene, Ig heavy locus (IGH@), it was upregulated by more than 10-fold in UC and 5.5-fold in CD against the control. They also reported that cadherin-11 (CDH11), decay accelerating factor for complement (DAF), mucin 1 (MUC1), phospholipase A2, group IIA (PLA2G2A), and tissue inhibitor of metalloproteinase 1 (TIMP1) were upregulated in both IBD subtypes.37 In this study, they also reported the discovery of an unknown gene, DKFZp547A023, which was subsequently confirmed by qPCR to be downregulated in both disease groups and it could serve as a potential candidate for further studies. As for the downregulated genes, their microarray results showed that cylindromatosis (CYLD), calcitonin gene-related peptide receptor component protein (RCP9), LIM protein (LIM), occluding (OCLN), Rho-associated, coiled-coil containing protein kinase 1 (ROCK1), and zinc finger, CCHC domain containing 4 (ZCCHC4) were involved in both IBD subtypes.37

Summary of the Transcriptomic Dysregulation Studies in IBD
Besides that, whole transcriptomic study on the identifications of aberrant RNAs also provides insightful correlations of other regulatory mechanisms which might be involved in the aetiopathogenesis of IBD. In a genome-wide cDNA microarray study by Palmieri et al.18 on the mucosal biopsies samples of 29 IBD patients (15 CD and 14 UC) has reported that out of 150 circadian genes, 21 genes (14%) in CD and 27 genes (18%) in UC showed upregulation, whereas 29 genes (19%) in CD and 23 (15%) in UC showed downregulation. Among the reported genes, ARNTL2, the core clock gene and RORA, nuclear hormones receptor were reported to be upregulated while PER3, the clock gene was downregulated in both conditions. As circadian clock circuitry is involved in the regulation of important cell processes and organ functions, changes in circadian cycle is involved in the basic mechanism of inflammatory and neoplastic diseases.18 It has also been suggested that dysregulated circadian cycle altered sleep patterns in UC and CD patients whereby this could affect the severity of the disease through the immune system activation and the release of inflammatory cytokines.63
Researchers were also trying to explore into the roles of lncRNAs in IBD, apart from focusing only on the mRNAs studies. For example, Mirza et al.3 had conducted a genome-wide study on lncRNAs and protein-coding genes profiling by microarray platform on inflamed and non-inflamed pinch biopsies from IBD patients (13 CD and 20 UC) and controls (n=12). They reported that there were 254 upregulated and 184 downregulated lncRNAs in inflamed CD and 370 upregulated and 375 downregulated lncRNAs in UC as compared to the control. Besides, there were 31 and 19 differentially expressed lncRNAs identified in the non-inflamed CD and UC against the control. The lncRNAs included RP11-731 F5.2, MMP12 and RP11-465 L10.10 were identified among the highly upregulated lncRNAs in both inflamed CD and UC against the control.3 DPP10-AS1, PDZK1P2 and antisense non-coding RNA in the INK4 locus (ANRIL) were reported to be downregulated while dual oxidase 2 (DUOX2) and its maturation factor, dual oxidase maturation factor 2 (DUOXA2), has been reported to be upregulated in association with inflamed UC.3 Their results concluded that the functional groupings of the most differentially expressed lncRNAs were enriched in the area of immune response and pro-inflammatory cytokine activity, suggested the involvement of this RNA in the persistent inflammation and pathogenesis of IBD.
A study on inflamed, non-inflamed and healthy mucosa layers of CD patients, Hong et al.21 had investigated the transcriptomic differences in terms of mRNA in between the cohorts in 13 CD patients (male, n=7) against 13 gender-match healthy individuals using RNA-sequencing (RNA-Seq) instead of microarray. About 950 genes were reported to be differentially regulated, 19 genes were detected to be significantly differentially expressed with chemokine (C-X-C) motif ligand 1 (CXCL1) gene reported to show the highest upregulation among them. Immunohistochemistry assay and qPCR further confirmed that CXCL1 gene expression was significantly steadily increased from no expression in normal mucosa to increased expression in inflamed CD mucosa. CXCL1 is a type of CXC chemokine which is known to be responsible in the accumulation of polymorphonuclear leukocytes for acute inflammation and infiltration of the inflamed tissue.21 Apart from being able to provide new findings on the roles of different RNAs in disease progression, the transcriptome studies on different RNAs (such as in mRNAs and lnRNAs) on a similar research backgrounds are important. This is because although the experiments were conducted by different groups, these results could be integrated and offer new perspectives on how the different RNAs interact and affecting the meta-omic environment of IBD.
In realizing the importance of understanding the complementation roles played by the different types of RNAs in meta-omic IBD research, recent experimental approach in etiopathology transcriptomic studies can be seen to adopt a more integrative model where the relationships of the paired expressions changes of different RNAs were investigated.62 A study by Palmieri et al. documented the integration of microarray expression profiles of ncRNAs and mRNAs in inflamed tissues of 15 CD patients relative to the normal mucosa. Additionally, they reported a more relevant and meaningful functional implications based on the paired co-expressions to the extent of pathway related to the disease through a computational statistical procedure. For example, they reported that miR-21, miR-126, miR-146a and miR-3194 possessed functional roles in CD as they were found to be differentially expressed and differentially co-expressed in the platelet activation signaling and aggregation pathway, one of the crucial dysregulated pathways in IBD. Through their analyses, they also discovered that some miRNAs that were differentially expressed did not involve in the differential co-expression with mRNA in the dysregulated biological processes pathways in IBD. On the other hand, some of the miRNAs that were not among the significantly differentially expressed list were found to be involved in the dysregulated pathways in IBD.
Other integrative experimental approaches such as metagenomic had also been reported in IBD studies. Häsler et al.61 investigated the relationship between the transcriptome changes in IBD patients with the microbiome of gut adherent mucosa-microbiota. The transcriptomic changes with regards to intestinal inflammation comparing between healthy and non-IBD inflamed control groups using NGS were determined in 63 biopsies (12 healthy, 15 disease control, 17 UC and 19 CD) in 41 individuals. Disease control groups were used to differentiate phenomenon to be IBD-specific and it is defined as infectious acute non-IBD inflammation. Intestinal microbiota has been known to play a role in shaping the immune response and other physiological functions in the intestinal environment. Any defective interactions between them could lead to the progression of IBD. Hence, Häsler et al.61 studied the host's transcriptomic responses and changes in the context of the microbiota signatures, inflammation and alternative splicing events in the development of the disease as they hypothesized that these could provide more implications on the understanding of the etiopathology of IBD rather than focusing on the level of differentially expressed genes alone. They discovered that there was no connection between gene expression of the host and the microbiota transcriptome in the inflamed colonic mucosa of IBD patients as compared to the non-IBD inflammation and healthy cohorts. Differential expressed genes were found to be involved in the pathophysiology of IBD such as interleukin-1 receptor type II, interleukin-6, and interleukin-8 while genes which showed alternative splicing were DUOX2, autophagy-related 16-like 1 and various interleukins. The most relevant pathway was microbiota-related inflammation pathway such as the chemokine signaling, natural killer cell-mediated cytotoxicity, NOD-like receptor signaling and spliceosome assembly.61
ADVANTAGES AND LIMITATIONS OF TRANSCRIPTOMIC STUDIES IN IBD
The reports of transcriptomic research in IBD are increasing as it provides evidence on the role of protein-coding mRNA and ncRNAs in modulating immune response in IBD.6465 In transcriptomic analysis, microarray and RNA-Seq techniques have been reported to be the current state-of-the-art and they had been widely utilized in studies with the effort to understand the etiology of IBD, especially on immune regulatory and signaling mechanisms.366 Furthermore, the high correlations between the high-throughput genome-wide association study (GWAS) data with independent transcriptomic data in IBD sample studies were reported to reveal the importance of gene expression signatures especially in inflammatory-related pathway.67 This had identified deregulated mRNAs and ncRNAs that are potentially utilized as biomarkers for the diagnostic of disease or served as targets in the discovery of novel therapeutic strategies. Nonetheless, there is also an example of study that showed the regulation of the pathways that are not detected by GWAS. A study by Cardinale et al.67 demonstrated that the downregulation in oxidative phosphorylation and upregulation of mitotic control pathways were detected in the gene expression analysis but not in GWAS study. Thus, although a huge amount of data could be obtained through GWAS study, independent transcriptomic studies are important to complement and validate the results obtained in GWAS related to IBD research. Moreover, the results from the transcriptomic study could only provide information on the putative identity of the aberrant RNAs.
MICROARRAY ANALYSIS VERSUS RNA-SEQUENCING IN TRANSCRIPTOMIC STUDY
Microarray and RNA sequencing are employed in most of the transcriptomic studies, especially in researches to investigate the molecular pathogenesis mechanism in human diseases. By looking at the summary of the discussed studies (Table 1), the earlier IBD transcriptomic studies were dominated by microarray analysis and utilization of the RNA sequencing technique only gained popularity in the recent years. Microarray analysis is a high-throughput simultaneous screening method of multiple targets arranged in a microchip while RNA sequencing is a process involving multiple stages including amplifications, sequencing, assembly and mapping from the whole transcriptome. While both techniques are commonly used to identify the transcript populations and their expression profiles in the sample, each of the techniques has their own strengths and weaknesses.
For the transcript identifications in microarray analysis, the arranged probes in the microchip are designed based on the currently available genomic sequences, whereas in RNA sequencing, each of the transcripts in the wh0ole transcriptome are sequenced and subsequently identified through reads assemblies and mapping.2166 Transcripts identification in the microarray analysis is restricted to the detection of transcripts with known genomic sequences while transcripts of both known and unknown (novel) sequences can be identified in the same sample source using RNA sequencing.21 Besides, it has been suggested that the results from RNA sequencing possesses higher reproducibility, better measurements of gene expressions level and additional information on isoforms can be provided as compared to the array method.216668 In RNA sequencing, specific read sequences of the splice junctions can be obtained and this leads to the direct identifications of known and also novel alternative splicing forms.
Performing RNA sequencing analysis is much more complicated than the microarray as there are more steps involved in the whole process. It would require a bioinformaticians or personnel experienced in bioinformatics to process and analyze the massive generated raw reads. The bioinformatics software often needs optimizations especially on the processes involved in analyzing the raw data. This includes processes like efficient data storage and retrieval, minimizing errors in image analysis, base calling and removal of low-quality reads before continuing with mapping and transcript assembly. While there are numerous tools developed and available to proceed with the analysis, the most suitable program has to be selected carefully depending on the project as different research questions cannot be answered and analyzed using similar bioinformatics tools.66
Microarray has been the commonly used technique to study transcriptomes in healthy condition or diseases before the advent of RNA sequencing. It is suitable to be used in expression profiling investigations with a defined pool of genes of interest, in performing targeted gene expression studies and in searching for selected alternatively spliced sequences. The volume of the data generated from the microarray analysis is high-throughput with relatively cheaper in term of cost per sample as compared to the RNA sequencing.66 In order to obtain a comprehensive representation of gene expression or for the detection of rare transcripts from complex organisms, repeated runs or more sequencing depth would be required in the sequencing of the samples. The need for repeated sequencing or more sequencing depth would imply a higher sequencing cost in RNA sequencing as compared to microarray analysis. In general, performing a RNA sequencing study would be more expensive and complicated than the microarray analysis and the most cost effective method should be decided based on the objectives of the experiment.
The problems that people face in microarray analysis relate with probe hybridization behavior, cross hybridization of similar sequence probes and in differentiating the signal-to-noise ratios even though custom arrays with specific probes of known sequences are used.68 The dependence of microarray analysis on known genomic sequences and the presence of high background noise due to cross-hybridizations had limited the detection depth of gene isoforms and alternative spliced transcripts species.66 Following this, the quantifications, detection sensitivity and calculated abundance of the transcripts could be compromised due to artefacts formations and subsequently affect the accuracy of genes expression level deteminations.68
FUTURE RESEARCH DIRECTIONS
Based on the current reported studies, transcriptome expression profiling studies had discovered numerous aberrant RNAs which are potentially involved in the pathogenesis of IBD. This information could be utilized as targets in the development of diagnostic biomarkers or therapeutic agents in IBD treatments. Microarray and NGS techniques are proven to be a useful tool in high-throughput genome-wide gene expression studies with verification studies using qPCR. More experimental designs incorporating transcriptomic studies could be conducted to further reveal on the molecular mechanisms and characterizations of RNAs in IBD by different study cohorts such as on the effects of disease duration of IBD patients leading to neoplasms. Knowledge on the changes in transcriptome could answer on how the different novel treatments could affect the progression of the disease. Functional validation studies are ought to be conducted to further confirm the roles played by these identified aberrant RNAs before incorporating them to any therapeutic treatments. While there have been several studies validating the functions of the aberrant RNAs in vitro,6970 there are relatively less functional studies being conducted in vivo.71 This is important as without knowing the functions of the discovered RNAs, targeted therapeutic approaches cannot be designed effectively. Although in vitro methods are commonly used in the functional validations, it is better to be complement with the more reliable in vivo validations using an animal models of IBD.72
CONCLUSIONS
High-throughput transcriptomic profiling techniques has become widely available serving as the common platform for data analysis and opportunities for the scientists to obtain a comprehensive understanding of IBD in term of genetic makeup. Transcriptomic analysis approach gained popularity due to their potentials towards identification of changes in the different RNA transcripts including novel and unknown transcripts underlying the IBD. More evidence-based researches will help to achieve a better understanding on the pathogenesis, etiology and complications of the complex and challenging disease such as IBD.
Notes
FINANCIAL SUPPORT: This research was supported from Fundamental Research Grant Scheme (FRGS)-FRGS/1/2015/SKK08/UKM/02/2 and Geran Universiti Penyelidikan (GUP)-GUP-2017-090.
CONFLICT OF INTEREST: No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION: Conceptualizations: SNC, NMM. Writing-original draft: SNC, NMM. Writing-review and editing: SNC, NMM, RARA. Approval of final manuscripts: all authors.
