Biosimilars: concept, current status, and future perspectives in inflammatory bowel diseases
Article information

Abstract
The inflammatory bowel diseases (IBD), which consist of Crohn’s disease and ulcerative colitis, are chronic, incurable immunemediated inflammatory disorders of the intestine. As IBD incidence continues to increase globally and its mortality is low, prevalent cases of IBD are rapidly increasing, thereby leading to a substantial increase in health care costs. Although the introduction of biologic agents for IBD management has revolutionized the armamentarium of IBD therapy, the high cost of this therapy is concerning. With the expirations of patents for existing biologic agents (originals), biosimilars with cheaper costs have been highlighted in the field of IBD. Despite concerns regarding their short- and long-term efficacy, safety, immunogenicity, and interchangeability, increasing evidence via prospective observations and phase III or IV clinical trials, which aim to prove the “biosimilarity” of biosimilars to originals, has partly confirmed their efficacy, safety, and interchangeability. Additionally, although patients and physicians are reluctant to use biosimilars, a positive budget impact has been reported owing to their use in different countries. In the near future, multiple biosimilars with lower costs, and efficacy and safety profile similar to originals, could be used to treat IBD; thus, further consideration and knowledge dissemination are warranted in this new era of biosimilars.
INTRODUCTION
Crohn’s disease (CD) and UC are 2 major subtypes of IBD that may cause chronic inflammation throughout the intestine, thereby resulting in hospitalization and surgery, which impair the quality of life of patients. IBD is an incurable immune-mediated chronic disorder with low mortality that requires lifelong management with anti-inflammatory agents, immunosuppressants, and biologic agents. In particular, the development and introduction of biologic agents with high efficacy and a good safety profile to IBD therapy have dramatically changed the treatment strategy of IBD therapy. In addition, the use of biologic therapy for the treatment of IBD has continuously increased during past decades in Western and Eastern countries [1-3].
The incidence of IBD continues to increase globally, including North America, the United Kingdom, Northern Europe, Australia, and recently developing regions such as Asia, the Middle East, and South America [4]. Currently, the prevalence of IBD in Western countries is up to 0.5% of the general population [5]. Because the incidence and prevalence of IBD continues to increase, the direct and indirect costs of IBD in Western countries are subsequently increasing. In the United States, direct medical costs related to IBD were more than USD 6 billion in 2004 [6,7]. The major health care costs in IBD management are driven by hospitalizations, surgery, outpatient visits, procedures, and pharmaceuticals. More importantly, the increasing use of biologic agents has led to a considerable rise in health care cost for IBD patients [8]. Currently, the patents of several biologic agents have either expired or are close to expiration, enabling the entry of copy versions of the original biologic agents, called biosimilars, into the market [9]. The introduction of biosimilars is expected to reduce health care costs and increase treatment access for patients with chronic, incurable immune-mediated disorders, such as IBD [10]. Because of the brief history of biosimilars, there are several debates regarding its biosimilarity, such as its efficacy, safety, immunogenicity, interchangeability, etc. to their originals. Therefore, in this review, we aimed to discuss the concept and the current status of biosimilars, and the future perspectives of biosimilars in the field of IBD on the global scale.
THE CONCEPT OF BIOSIMILAR
Although biosimilars are known as highly similar agents to the originals (the reference biologic products), they should not be referred to as, nor equated with, generic drugs, which are equivalent to original products in several aspects such as dosage, strength, route of administration, performance, etc. There should be no clinically meaningful differences between biosimilars and their originals based on safety, purity, or potency [11]. Because biologic agents are synthesized in living cells, 2 biologic drugs cannot be structurally identical due to unique posttranslational modifications. Of note, the scientific principles underlying comparability that are applicable to biosimilars are the same as those related to changes in the manufacturing process of the originals. For example, since the first approval of infliximab (Remicade®; Janssen Biotech, Inc, Horsham, PA, USA) in 1999, there have been more than 35 changes in the manufacturing process for its production, including process improvements, scale changes, and site transfers. However, acceptable product quality and comparability have always been demonstrated during these changes [12,13].
REGULATORY PATHWAY FOR BIOSIMILARS
The regulatory process for the approval of biosimilars differs from that of original drug products [14,15]. For new biologic agents (i.e., originals), they must be tested in each clinical indication for which approval is sought; this step is usually the most timeconsuming and costly phase of new biologic development. In contrast, for biosimilars, an abbreviated approval process can be used, which mainly focuses on its structural, analytical, and in vitro similarity to the reference product. Following this pathway, only one comparative clinical trial that includes assessment of pharmacokinetics and immunogenicity is required before the approval of biosimilars and these data can be extrapolated to other indications. Through this pathway, the first biosimilar to infliximab, CT-P13 (Remsima; Celltrion, Incheon, Korea and infliximab-dyyb, Inflectra®; Pfizer, New York, NY, USA), was approved by the European Medicines Agency (EMA) in 2013 and the US Food and Drug Administration (FDA) in April 2016 [16,17]. The use of CT-P13 for IBD indications was extrapolated from the results of randomized clinical trials (RCTs) in rheumatoid arthritis (RA) and ankylosing spondylitis (AS) [18,19]. To date, a growing body of evidence from several retrospective and pharmacovigilance data continue to support the proposal that the efficacy and safety of CT-P13 is comparable to those of the original drug [20-22].
EFFICACY AND SAFETY IN PROSPECTIVE STUDIES
The first prospective, observational study assessing the efficacy, tolerability, and safety of CT-P13 was presented by Jahnsen et al. in 2015 [23]. From January 2014 to February 2015, a total of 78 IBD patients (46 with CD and 32 with UC) were recruited, and 30% of CD and 25% of UC patients were previously treated with anti-TNF agents. In CD, 79% of patients (34/43) achieved clinical remission, which was defined as a Harvey-Bradshaw index score ≤ 4 at week 14. In UC patients, the remission rate, defined by the partial Mayo score ≤ 2, was 56% (18/32) at week 14. During the study period, there were no unexpected adverse events. In 2017, Gecse et al. [24] reported the final results from a prospective, multicenter, nationwide Hungarian IBD cohort, including 353 consecutive patients receiving CT-P13 (209 with CD and 144 with UC), of whom 229 patients reached the 54-week endpoints. In patients with CD, clinical remission was achieved in 49%, 53%, and 48% of patients, while clinical response was observed in 86%, 81%, and 65% of patients, at weeks 14, 30, and 54, respectively. In UC, clinical remission was detected in 56%, 41%, and 43% of patients, while clinical response was observed in 74%, 66%, and 50% of patients, at weeks 14, 30, and 54, respectively. Patients who had previously received anti-TNF therapy showed significantly lower remission and response rates at weeks 14, 30, and 54 for both CD and UC. Infusion reactions were reported in 8.8% of patients, while infections were reported in 9% of patients; 1 death was also observed. There were no new safety signals from this cohort of patients. Recently, the largest experience with CT-P13 was demonstrated via an Italian prospective, nationwide, multicenter, observational cohort, named PROSIT, which evaluated its safety, and its clinical and endoscopic efficacy [25]. These researchers recruited 680 consecutive IBD patients (373 with CD and 307 with UC) from 25 sites and mean follow-up was 32 weeks. This heterogeneous cohort of patients comprised 400 patients naive to anti-TNF therapy, 171 patients who were previously exposed to biologic agents, and 109 patients who were switched from originator infliximab to CT-P13. Although primary failure of induction therapy was observed in 8.1% of patients, as a whole, 45.6% (274/601) of patients were in remission, 30.9% (186/601) were deemed responders, and 10.3% (62/601) experienced loss of response during the follow-up without new safety signals. Additionally, deep remission was achieved in 57% of CD and 50% of UC patients (Table 1).
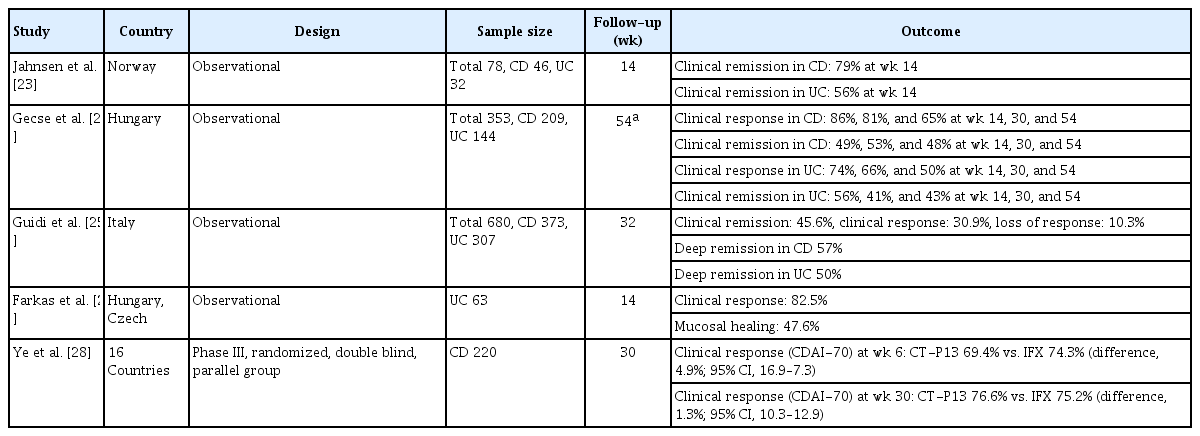
Prospective Studies for the Clinical Efficacy of CT-P13 in IBD
The efficacy of CT-P13 induction therapy on mucosal healing in patients with UC was evaluated by Farkas et al. [26] in a prospective, multicenter Hungarian and Czech study. Altogether, 63 consecutive UC patients receiving CT-P13 therapy due to acute, severe relapse (24 patients) or for chronic refractory disease activity (39 patients) were included. Cumulative clinical response and steroid-free remission at week 14 were achieved in 82.5% and 47.4% of patients, respectively. Mucosal healing, which was defined as an endoscopic Mayo score of 0 or 1, was detected in 47.6% of patients, and complete mucosal healing (i.e., endoscopic Mayo score 0) was found in 27% of patients. RCTs of the originator infliximab resulted in clinical response, remission, and mucosal healing rates of 69%, 39%, and 62%, respectively, at week 8 in Active Colitis Trials (ACT) 1 and 65%, 34%, and 60% at week 8 in ACT 2 [27]. Compared to the ACT 1 and 2 results, the higher response and remission rate and the comparable rate of mucosal healing in this study could be explained by the longer term of outcome evaluations in this study (week 14 vs. week 8), and may support the notion that real-life data depict better outcomes than clinical trials.
Recently, Ye et al. [28] reported the results from a phase III randomized, double blind parallel group trial, conducted in patients with moderate to severe CD. These authors compared the efficacy of CT-P13 to originator infliximab, with 220 patients from 16 countries. The primary outcome of this study was to compare the efficacy between 2 groups based on CDAI-70 response rates (defined as a reduction from the baseline CDAI score by at least 70 points) at week 6. In a per protocol analysis, CDAI-70 response rate was quite similar (CT-P13 69.4% vs. originator 74.3%: difference, 4.9%; 95% CI, 16.9–7.3) and based on the significant clinical response (CDAI-100 response, decrease in CDAI score of at least 100 points from baseline), no difference was observed (CT-P13 60.4% vs. originator 64.2%: difference, 3.9%; 95% CI, 16.7–9.6). Week 30 data also revealed similar results between the 2 groups based on CDAI-70 response rate, CT-P13 76.6% versus originator 75.2%; CDAI-100 response rate, CT-P13 72.1% versus originator 73.4%; and clinical remission (CDAI score of less than 150 points) rate, CT-P13 55.0% versus originator 56.9%. In this randomized study, CT-P13 was well tolerated and displayed a safety profile comparable to that of originator up to 30 weeks. The result of this phase III study may help overcome the concern regarding the extrapolation issue related to biosimilars.
SWITCH FROM THE ORIGINAL TO THE BIOSIMILAR, INFLIXIMAB
The NOR-SWITCH Study was a 52-week, randomized, noninferiority double blind trial that examined switching from reference infliximab to CT-P13 and its efficacy, safety, and immunogenicity [29]. This study was financed by the Norwegian Government and monitored using the health care system. Adult patients on stable treatment with reference infliximab and treated in the hospital settings for at least 6 months were recruited to participate in this study. The primary endpoint was disease worsening during a 52-week follow-up period. Overall, 482 patients across multiple indications were enrolled and randomized in a 1:1 ratio (241 to reference infliximab and 241 to CT-P13). Patients suffered from CD (32%, 155 patients), UC (19%, 93 patients), and the remaining 233 patients (49%) suffered from other diseases, including AS (19%), RA (16%), psoriatic arthritis (6%), and chronic plaque psoriasis (7%). Across all indication, disease worsening was detected in 26% (53 patients) on originator infliximab and 30% (61 patients) of patients in the CT-P13 group (adjusted treatment difference, 4.4%; 95% CI, 12.7–3.9). The frequency of serious adverse events was 10% in the reference group and 9% in the CT-P13 group, and the same frequency of adverse events led to therapy discontinuation (4% vs. 3%, respectively). The authors concluded that switching from originator infliximab to CT-P13 was not inferior to continued treatment with originator, according to a pre-specified non-inferiority margin of 15%. Although this study was not powered to demonstrate non-inferiority in individual diseases, authors performed an explorative subgroup analysis in 129 CD patients and 75 UC patients, and the result showed similarity between patients treated with originator infliximab and CT-P13 based on efficacy, safety, and immunogenicity (Table 2) [30].

Prospective Studies for CT-P13 Switching in IBD
Another experience of switching was summarized by Kolar et al. [31] from one tertiary site in the Czech Republic. In this study, there were 2 cohorts of patients: cohort 1, 74 IBD patients (56 with CD and 18 with UC) who were switched from originator infliximab to CT-P13; and cohort 2, 119 infliximab-naive patients (90 with CD and 29 with UC) who were newly administered CT-P13. In the switching cohort, most patients remained stable, and the remission rate at weeks 0 and 56 were 72.2% versus 77.8%, respectively. The median difference between both Harvey-Bradshaw index scores in CD and Simple Clinical Colitis Activity Index scores in UC between weeks 0 and 56 was 0. Additionally, there were no significant differences in CRP (4.3 ± 8.0 mg/L vs. 3.3 ± 3.8 mg/L; P= 0.82) or fecal calprotectin (135 ± 153 μg/g vs. 199 ± 225 μg/g; P= 0.84). In the infliximab-naive cohort, 92% and 86% of CD patients and 83% and 64% of UC patients responded to CT-P13 at weeks 14 and 46, respectively. Moreover, half of the UC patients experienced mucosal healing at week 14, and 95% of CD patients experienced improvement in perianal disease at week 46. In addition, there was no increase in immunogenicity in switched patients (antidrug antibody [ADA] positivity 9.5% at switch vs. 6.0% at week 54; P= 0.54). The types and frequency of adverse events were comparable to originator infliximab in both cohorts. During the follow-up, CT-P13 was discontinued in 7 patients with CD due to serious adverse events, including 2 patients from the switching group (1 with delayed allergic reaction and 1 with fibrous dysplasia located in maxilla) and 5 patients from the naive group (2 patients with skin complications including abscesses and rash, 2 patients experiencing lupus-like joint symptoms, and 1 patient with palmoplantar pustulosis). Razanskaite et al. [32] reported a switching experience from the United Kingdom where 143 IBD patients were switched from originator infliximab to CT-P13. Patients reported a similar incidence in side effects before and after the switch. There was no significant change in drug persistence between patients treated with CT-P13 in the preceding year and those treated with originator infliximab (P= 0.94).
In 2017, recommendations regarding the interchangeability between originals and biosimilars were announced in Europe and the United States. The updated position of the European Crohn’s disease and Colitis Organisation (ECCO) on biosimilars stated that the usage of biosimilars is increasing and it plays an important role in the treatment of IBD patients. In addition, the report confirmed that switching from the originals to biosimilars is acceptable [33], but should be performed after careful discussion among physicians, nurses, pharmacists, and patients. The American Gastroenterological Association has recommended that prescribing physicians should prevent nonmedical switching from originals to biosimilars [34].
IMMUNOGENICITY AND PHARMACOKINETICS
Antibodies to biologic agents are known as a risk factor for the development of acute infusion reactions and loss of response, which were reported in 5% to 23% of patients treated with originator infliximab [35]. Patients who developed antibodies against originator infliximab had a 2-fold risk of acute infusion reactions and a 6-fold risk of serious acute infusion reactions [36]. The incidence of infusion reactions was 16% and 21% in ACCENT 1 and 2 studies (RCTs of originator infliximab in CD), respectively, where the lowest incidence occurred in patients receiving corticosteroids and immunosuppressants. Moreover, in the ACT 1 and 2 studies, infusion reactions were observed in 9.9% and 11.6% of patients, respectively [27,37,38]. In the 2 pivotal trials (PLANETAS and PLANETRA) comparing CT-P13 to originator infliximab in patients with AS and RA, infusion reactions were observed in 3.9% and 4.9% of patients with AS, and 6.6% and 8.3% of RA patients for CT-P13 and originator infliximab, respectively [18,19]. A phase III study comparing CT-P13 with originator infliximab in CD showed no difference in the rate of infusion reactions between both groups at week 30 (CT-P13 7.2% vs. originator infliximab 8.3%) [28].
Bálint et al. [39] focused on the immunogenicity of CT-P13 in a central European cohort, including 384 consecutive patients (253 CD and 131 UC; 291 Hungarian and 93 Czech) from 13 Hungarian and 1 Czech tertiary IBD sites. Mean CT-P13 trough levels (TLs) were 20.1, 14.7, and 5.0 μg/mL at weeks 2, 6, and 14, respectively, and cumulative rates of ADA positivity were 8.7%, 19.3%, and 28.0% at weeks 0, 14, and 30, respectively. During induction and maintenance treatment, infusion reactions were observed in 7.3% of patients (28/384), and were most frequent with the 2nd and 3rd infusions. All infusion reactions were mild to moderate, and the most common symptoms of infusion reactions were flushing, dyspnea, and chest discomfort. CT-P13 therapy had to be terminated in 17 patients, who were switched to adalimumab in 12 cases. The results of this study also suggested comparable rates and characteristics of infusion reactions between CT-P13 and originator infliximab.
In the interim report of a prospective nationwide study published by Gecse et al. [40], mean TLs of CT-P13 did not differ between CD and UC patients, except for week 6 results, where TLs were significantly higher in CD patients than UC patients (18.4 μg/mL vs. 6.2 μg/mL; P< 0.001). Patients who had been previously exposed to an anti-TNF agent had lower TLs than naive patients (15.0 μg/mL vs. 21.5 μg/mL at week 2; 7.7 μg/mL vs. 10.2 μg/mL at week 6; and 4.8 μg/mL vs. 3.2 μg/mL at week 14, not significant). ADA positivity was detected in 9.1% and 21.3% of CD patients and 8.8% and 23.8% of UC patients at weeks 0 and 14, respectively. In the same study with an enlarged cohort of 291 patients [41], patients with previous originator infliximab exposure had significantly higher ADA positivity in both CD and UC. In this analysis, the overall ADA positivity was 28% at week 30 (24.1% in naive patients and 44.4% in previous anti-TNF exposed patients), and concomitant azathioprine (AZA) therapy prevented early ADA formation in antiTNF naive patients in both CD and UC (AZA group 11.2% vs. non-AZA group 24.6%, P= 0.012); however, these effects were lost at week 30. In the Norwegian study presented by Jahnsen et al. [23], which included 78 patients, median TLs at week 14 were 6.8 μg/mL in CD and 6.2 μg/mL in UC patients. TLs were not detected in 8 patients (4 with CD and 4 with UC), 3 of whom had previously received anti-TNF therapy. Two of the 8 patients had high ADA levels while 5 patients had medium/high levels. In a multicenter central European study focused on mucosal healing induced by CT-P13 in UC after an induction period, a significant correlation between TLs of CT-P13 and clinical and endoscopic responses was observed, with a cutoff value of 3.15 μg/mL in the ROC analysis for both steroid-free remission and mucosal healing [26].
Recently, Ben-Horin et al. [42] examined the presence of crossimmunogenicity of CT-P13 with originator infliximab. This in vitro study revealed that antibodies which had developed against originator infliximab during previous exposure to the drug were also cross-reactive with CT-P13. This important finding suggests that similar immunogenicity and immunodominant epitopes are shared between these infliximab agents (originator infliximab and CT-P13). In a Czech switching cohort of IBD patients treated with CT-P13, no difference in ADA positivity (9.5% vs. 6.0%, P= 0.54) and an increase in TLs (3.4 ± 3.8 μg/mL vs. 4.7 ± 4.5 μg/mL, P= 0.01) were observed at switching and week 56 post-switching from originator infliximab [31]. In another switching study performed by Smits et al. [43,44], median TLs obtained during the maintenance phase increased significantly from week 0 to 16 (3.5–4.2 μg/mL, P= 0.010), and the proportion of patients with TL within the therapeutic range (3.0–7.0 μg/mL) increased from 39% (week 0) to 45% (week 52). Of the 83 patients, 7 (8%) developed ADA during the follow-up period and 5 had preexisting detectable ADA levels at baseline. No increase in immunogenicity was observed after switching to CT-P13 in pediatric IBD patients in a study by Sieczkowska et al. [45] who assessed 16 and 15 patients with CD at the time and after switching from originator infliximab to CT-P13, respectively. At switch, 14 out of 16 children had therapeutic TLs and 7 patients had positive ADA levels. After switching, 15 out of 15 patients had therapeutic TLs and only 4 patients developed ADA positivity. Strik et al. [46] also presented the preliminary results of the SECURE Study which was performed to prospectively evaluate serum drug concentration 16 weeks after switching from originator infliximab to CT-P13 in subjects in stable remission. In 44 CD patients, mean serum TLs were 2.97 μg/mL before switching and 3.25 μg/mL at 16 weeks after switching, thereby demonstrating the non-inferiority of CT-P13 to originator infliximab.
ECONOMIC ISSUES
The introduction of new, expensive biologic agents is associated with an increase in the direct medical costs of IBD management. Biologic therapy has thus been less accessible in countries with low and middle gross national product (GNP) per capita. Biosimilars could enable health economic savings and improve the access of patients to biologic therapy as they are priced lower than the originals but demonstrate highly comparable efficacy, safety, and immunogenicity profile to their proprietary counterparts. It is predicted that by 2020, biologic agents will form 28% of the global pharmaceutical market by value, with biosimilars offering potential savings greater than 50 billion EUR across the European “Big 5” countries and the USA [47]. Cost savings is considered to be the main advantage of biosimilars for IBD patients. By performing web-based surveys regarding biosimilars in 2013 and 2016, the ECCO found that cost saving was thought to be the main advantage of biosimilars by approximately 90% of respondents [48,49]. These savings can be reinvested to treat additional patients with IBD or be applied to other segments, including patients and health care professional (e.g., IBD nurses) education. However, the relative price reduction of biosimilars might not be as profound as that of chemical generics, where competition between multiple manufacturers finally drives prices in a downward direction to more than 50% in most markets [50,51]. Because of the considerable costs associated with manufacturing, marketing, storage, and special requirements for pharmacovigilance, a greater time might be required to drive the cost savings effect of biosimilars until more biosimilars enter the market to cause a high level of market competition [50]. Nonetheless, a massive price reduction for biosimilars has been observed in various countries. In Norway, CT-P13 enabled a cost reduction of 51%–65% compared to originator infliximab; in France, the price discount is 45%, and in Japan, the current price of CTP13 is 67% lower than that of originator infliximab [52]. Thus, the magnitude of price reduction may be important for switching prescriptions to biosimilars. Kanters et al. [53] performed a budget impact analysis using a Delphi panel for the adoption of the biosimilar, infliximab, for rheumatologic disorders and IBD in 5 European countries. In this study, savings were expected in all countries for all diseases, when larger price reductions (50% or more) of the biosimilar infliximab were performed (i.e., physicians would prescribe biosimilars only when price reductions were sufficiently large). Another survey from the United States showed that of 150 physicians, 83% would prescribe biosimilars if they were 25% cheaper and would only prescribe biosimilars to naive patients instead of patients currently or were previously treated with the original [54].
Brodszky et al. [55] presented a very interesting budget impact analysis that focused on 6 lower- and middle-income European countries, with the assumption of a 25% price reduction. In the first scenario, 8 million EUR in saving was achieved in these countries if only new IBD patients were treated with CTP13 for a 3-year period (i.e., switching was not allowed in this scenario). In the second scenario, where 80% of IBD patients received CT-P13 (switching was allowed), 16.9 million EUR in savings was achieved. Most importantly, by saving money, 722 to 1,530 additional IBD patients could be treated with CT-P13. Jha et al. [56] focused on the budget impact analysis model in 5 higher income European countries and assumed price reductions of 10%, 20%, and 30%, respectively. The expected cost savings increased from 11.9 million EUR to 35.8 million EUR in CD and 5.1 million EUR to 15.4 million EUR in UC. If these saving budgets were used to treat additional IBD patients, 1,219–4,701 additional patients could be treated in those 5 countries for 1 year. In the Czech Republic, the rapid and robust reduction (30%–40%) in the cost of biosimilars has facilitated earlier initiation of biologic therapy in IBD patients on treatment waiting lists, with an additional 1,000 patients receiving treatment in 2014 compared to the previous year [57]. In the switching experience from the United Kingdom, where 143 IBD patients switched from originator infliximab to CTP13 according to Razanskaite et al. [32], drug acquisition costs decreased by 40,000–60,000 pounds per month following the initiation of the switching program.
Recently, Rencz et al. [58] published a probabilistic Markov model comparing the cost-effectiveness of treatment sequences with available biologics, including adalimumab, CTP13, originator infliximab, and vedolizumab, for luminal CD in 9 European countries. In addition, they applied the countryspecific unit costs, discount rates, and a third-party payer perspective to the model. Compared to conventional therapy, CTP13 resulted in the most favorable incremental cost utility ratios, which ranged from 34,580 EUR/quality-adjusted life year (QALY) in Hungary to 77,062 EUR/QALY in Sweden. Based on the calculated results, CT-P13 was recommended as a firstline biologic treatment for luminal CD that is unresponsive to conventional therapy. Baji et al. [59] reported the cost-effectiveness model of different biologic sequences including originator infliximab, CT-P13, adalimumab, and vedolizumab in 9 European countries for fistulizing CD. CT-P13 was the most cost-effective drug against the standard care across countries, with incremental cost-effectiveness ratios between 34,684 EUR/QALY and 72,551 EUR/QALY. According to this model, the first choice of therapy in fistulizing CD was CT-P13 and when treatment fails, switching to adalimumab and then to vedolizumab provides the meaningful health benefits required, but at an increased cost [59].
THE PERSPECTIVES OF PHYSICIANS AND PATIENTS
In 2013 and 2016, the ECCO performed 2 surveys with its members to determine their knowledge of and attitude toward biosimilars [48,49]. The first survey, performed in 2013 (the year CTP13 was approved by the EMA in Europe), showed that physicians had minor confidence in the use of biosimilars. In addition, lower cost was identified to be the major advantage of biosimilars. However, respondents had concerns regarding the extrapolation of data across indications, and the immunogenicity, safety, and interchangeability of biosimilars. Following more extensive use of biosimilars across Europe and the growing body of evidence regarding their biosimilarity to respective originals, a second survey was performed in 2015. This survey also revealed that cost saving was considered to be the main advantage of biosimilars by most respondents (92%), and immunogenicity was the main concern (69% of respondents). Furthermore, the biosimilars were considered to be interchangeable with the proprietary products by 44% of respondents, which increased relative to that found in the 2013 survey (only 6%). Nonetheless, 90% disagreed with the automated substitution by dispensing pharmacists, and only 33% were against extrapolation across indications compared to 76% in 2013. The survey also revealed that only 25% of the respondents did not favor the extrapolation of data across different subtypes of IBD, compared to 53% in 2013. Such findings suggest an increase in confidence regarding the use of biosimilars, with only 20% of respondents having little or no confidence in their use, compared to 63% in 2013.
A survey from Germany revealed that some patients were reluctant to accept a biosimilar prescription. In addition, this reluctancy was particularly identified among patients who were currently receiving the proprietary medicine and were not displaying any clinically-indicated reasons that required a switch to the biosimilar [60]. The European Federation of Crohn’s and Ulcerative Colitis Association carried out a survey to investigate patients’ perspectives regarding biosimilars [61]. Between 2014 and 2015, 1,181 patients responded to the survey, but only 38% had prior knowledge of biosimilars and only 25% had no concerns regarding their use. In contrast, 47% and 40% of the respondents expressed their concern regarding its safety and efficacy, respectively, while 35% were concerned that their molecular basis might differ from the brand-name drugs. More than half of the respondents expressed that cost savings should not precede safety and efficacy, and only 31% were fully confident in biosimilars, even when they were prescribed the biosimilar and provided with an explanation by the treating physician. This survey also revealed that patients wished to be informed and involved in the decision-making process regarding biosimilar dispensing; 66% preferred to be told which drug they are prescribed (i.e., original or biosimilar), and 21% rejected the concept of interchangeability if the patient was not aware. Based on these findings, communication between patients and physicians is important during the course of IBD management.
UNRESOLVED ISSUES
Several biosimilars, besides CT-P13, are currently in preclinical and clinical trials worldwide [62]. Also, several infliximab biosimilars other than CT-P13 have been approved by FDA or EMA from 2016 to 2019; Renflexis (Samsung Bioepis), Ixifi (Pfizer), and Avsola (Amgen) are approved by FDA. Zessly (Sandoz) and Flixabi (Samsung Bioepis) are approved by EMA. In addition, there are several adalimumab biosimilars approved by FDA or EMA so far; Amjevita (Amgen), Cyltezo (Boehringer Ingelheim), Hyrimoz (Sandoz), Hadlima (Samsung Bioepis), and Abrilada (Pfizer) are approved by FDA. Kromeya (Fresenius Kabi), Idacio (Fresenius Kabi), Hulio (Mylan S.A.S), Hyrimoz (Sandoz), Hefiya (Sandoz), Halimatoz (Sandoz), Imraldi (Samsung Bioepis), and Amgevita (Amgen) are approved by EMA. In fact, they are either ready to enter or have already entered the market [63,64], although there has been a lack of data regarding biosimilar experiences in IBD with them except a few published studies [65,66]. Also, there still remains a lack of scientific and clinical evidence regarding reverse switching, multiple switching, and cross-switching among biosimilars for IBD patients. Furthermore, automated substitution by the dispensing pharmacists would serve as an additional issue in the era of multiple biosimilars for IBD indications [33]. Therefore, the extended (i.e., an even longer term) safety of biosimilars should be monitored through formal postmarketing observational studies [34].
CONCLUSION
Based on the current findings regarding the efficacy and safety of biosimilars in the field of IBD and the decreased reluctance to use biosimilars by patients and physicians, we recognized that a growing desire exists to cut health care costs and use the savings to invest in and assist more patients. Achieving these measures would ultimately enable the creation of a virtuous cycle where future health care costs are decreased. Several prospective studies are currently ongoing and their results could help to confirm the long-term efficacy and safety of biosimilars, and clarify ongoing debates regarding several unresolved or partially resolved issues, such as extrapolations, switching, and the immunogenicity of biosimilars. As the era of biosimilars is presently unavoidable, gaining a better understanding of biosimilars, especially for near future applications, is a crucial requirement in the field of therapeutics.
Notes
FINANCIAL SUPPORT
The authors received no financial support for the research, authorship, and/or publication of this article.
CONFLICT OF INTEREST
Doctor Loftus EV Jr. has consulted for AbbVie, Allergan, Amgen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Celltrion Healthcare, Eli Lilly, Genentech, Gilead, Janssen, Pfizer, Takeda, and UCB; and has received research support from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Genentech, Gilead, Janssen, Pfizer, Robarts Clinical Trials, Takeda, and UCB. No other potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION
Conceptualization: Park SH, Park JC, Loftus EV Jr. Writing - original draft: Park SH, Park JC, Loftus EV Jr. Writing - review and editing: Lukas M, Kolar M. Approval of final manuscript: all authors.