Efficacy and safety of ustekinumab in Japanese patients with moderately to severely active Crohn's disease: a subpopulation analysis of phase 3 induction and maintenance studies
Article information
Abstract
Background/Aims
Efficacy and safety of ustekinumab were evaluated in a Japanese subpopulation with moderately to severely active Crohn's disease (CD) in UNITI-1, UNITI-2 and IM-UNITI studies and results were compared with the overall population.
Methods
Overall, patients in UNITI-1 (Japan, n=56; failed response to tumor necrosis factor antagonist) and UNITI-2 (Japan, n=26; failed response to prior conventional therapy) were randomized to placebo or ustekinumab intravenous induction (130 mg or ~6 mg/kg) at week 0. Responders to ustekinumab induction therapy (Japan, n=21) were randomized to placebo or ustekinumab (90 mg, subcutaneous) maintenance (every 12 weeks [q12w] or 8 weeks [q8w]) in IM-UNITI. The primary endpoint was clinical response at week 6 for induction studies and clinical remission at week 44 for maintenance study.
Results
Percentage of patients achieving clinical response at week 6 was greater in ustekinumab 130 mg and ~6 mg/kg groups than in the placebo group (UNITI-1: 36.8% and 31.6% vs. 27.8%, respectively, for Japanese; 34.3% and 33.7% vs. 21.5%, respectively, for overall; UNITI-2: 37.5% and 55.6% vs. 11.1%, respectively, for Japanese; 51.7% and 55.5% vs. 28.7%, respectively, for overall). Clinical remission rate at week 44 during maintenance was greater in the ustekinumab 90 mg SC q12w and q8w groups than in the placebo group (50.0% and 55.6% vs. 25.0%, respectively, for Japanese; 48.8% and 53.1% vs. 35.9%, respectively, for overall). Efficacy and safety results observed in the Japanese subpopulation were generally consistent with those in the overall population.
Conclusions
Ustekinumab could be considered as a new therapeutic option for moderately to severely active CD in Japanese patients. Both ustekinumab induction and maintenance treatments were generally well tolerated (Clinical Trial Registration: NCT01369329, NCT01369342, NCT01369355).
INTRODUCTION
Crohn's disease (CD) is a chronic, immune-mediated IBD that is often characterized by remission and recurrent relapses.12 The incidence of CD has increased over the past 5 decades in developed countries, and it varies from 0 to 20.1 cases per 100,000 persons in North America and from 0.3 to 12.7 cases per 100,000 persons in European countries.3 The incidence of CD in Japan has increased from 0.60 to 1.20 between 1986 and 1998, and although it is much lower than in the Western countries, it has rapidly increased in the past 20 years.45 The changing dietary habits due to urbanization and other environmental risk factors along with the changes in gastrointestinal microbiota may be responsible for the rising incidence of CD in Japan.678
The current standard of care for CD in Japan consists of corticosteroids for induction and 5-aminosalicylic acid, azathioprine, 6-mercaptopurine, and tumor necrosis factor (TNF) antagonists (infliximab and adalimumab) for induction and maintenance.910 TNF-antagonists are the only biologic therapies available for patients with moderately to severely active CD in Japan. Although TNF antagonists have advanced the management of CD, a considerable number of patients show primary nonresponse, loss of response, or intolerance to the treatment.11121314 Therefore, there is currently an unmet need for additional treatment options with new mechanisms of action for moderately to severely active CD, particularly in Japan.
Ustekinumab is a fully human monoclonal IgG1 antibody that targets the biological activity of interleukin (IL)-12/23 through their shared subunit p40 and inhibits signaling through these 2 cytokine receptors.15 Ustekinumab is currently approved for the treatment of psoriasis and psoriatic arthritis in various countries, including Japan,1617 and it is currently approved for the treatment of moderately to severely active CD in the United States and Europe. In a previous phase 2b global study conducted in moderately to severely active CD patients who were refractory to TNF antagonists, intravenous (IV) ustekinumab induction therapy showed increased rates of clinical responses at week 6 (36.6% for 1 mg/kg, 34.1% for 3 mg/kg, and 39.7% for 6 mg/kg vs. 23.5% for placebo, P=0.005), and SC ustekinumab maintenance therapy showed increased rates of response (69.4% vs. 42.5%, P<0.001) and remission (41.7% vs. 27.4%, P=0.03) at week 22.18 A phase 3 global clinical study program, comprising of two 8-week induction studies (UNITI-1 and UNITI-2) and a 44-week maintenance study (IM-UNITI), was designed to evaluate efficacy and safety of ustekinumab over a period of 52 weeks in patients with moderately to severely active CD who were refractory to TNF antagonists or conventional therapy.19 Here, we report the efficacy and safety of ustekinumab from the UNITI-1, UNITI-2, and IM-UNITI global studies in a Japanese subpopulation.
METHODS
1. Study Overview
The phase 3 ustekinumab development program for CD comprised of two 8-week induction studies (UNITI-1 [NCT01369329] and UNITI-2 [NCT01369342]) and one 44-week randomized withdrawal (of ustekinumab-induction responders) maintenance study (IM-UNITI [NCT01369355]) (Fig. 1). The details on the UNITI-1, UNITI-2, and IM-UNITI study designs have been described elsewhere.19 All the 3 studies were multicenter, randomized, double-blind, placebo-controlled, and were conducted between July 2011 and June 2015 in various countries, including Japan (13 sites for UNITI-1 and 16 sites for UNITI-2, with subjects from 20 sites continuing to participate in IM-UNITI).
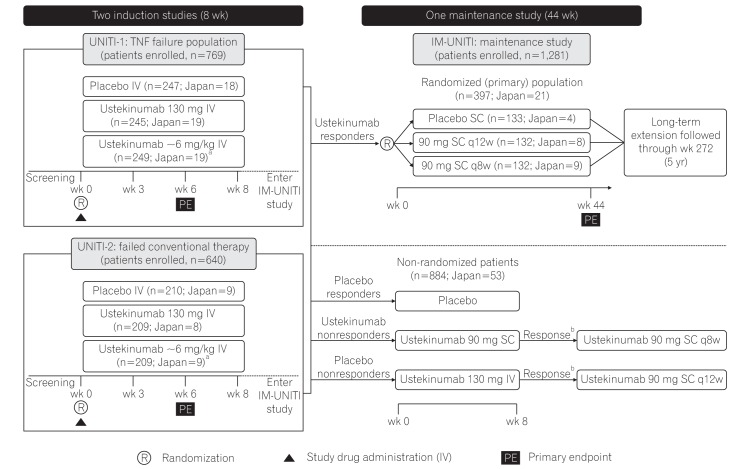
Study design of phase 3 CD program for ustekinumab. Patients randomized to placebo and patients who were nonresponders to ustekinumab were eligible for nonrandomized maintenance dose after completion of induction study. aWeight range based ustekinumab doses approximating at 6 mg/kg; bIf there was no clinical response achieved at week 8, the study treatment was discontinued. TNF, tumor necrosis factor; IV, intravenous; q8w, every 8 weeks; q12w, every 12 weeks. Adapted from Feagan BG, et al. N Engl J Med 2016;375:1946-1960.19
Patients who were ≥18 years of age with ≥3 months of CD with a CDAI score of 220 to 450 were eligible for induction studies. In UNITI-1 study, patients were required to have had primary or secondary nonresponse or intolerance to ≥1 TNF antagonists. In UNITI-2, patients were required to have failed conventional therapy, and they could have undergone TNF antagonist(s) therapy but should not have demonstrated a primary or secondary nonresponse or intolerance. Treatment with immunosuppressants, mesalamine, antibiotics, and/or oral corticosteroids (prednisone ≤40 mg/day or budesonide ≤9 mg/day) at stable doses was permitted. Patients previously treated with IL-12 or IL-23-antagonists were excluded. Patients with Crohn's complications anticipated to require surgical treatment or that could impede the assessment of treatment response using CDAI, active tuberculosis, and other infections or history of cancer were also excluded. Eligible patients in both induction studies were randomized to receive a single IV dose of placebo or ustekinumab (130 mg dose) or a weight-based dose of ustekinumab approximating 6 mg/kg (260 mg [weight ≤55 kg], 390 mg [weight >55 kg and ≤85 kg], or 520 mg [weight >85 kg]) at baseline (week 0).
Patients who responded to ustekinumab IV at week 8 in either of the induction studies were eligible to enter the 44-week maintenance study (IM-UNITI) as the primary (randomized) population. These patients underwent randomized withdrawal, receiving SC administration of either placebo or 1 of 2 ustekinumab maintenance regimens (ustekinumab 90 mg every 12 weeks [q12w] or ustekinumab 90 mg every 8 weeks [q8w]). Patients who did not show a clinical response to ustekinumab induction and all placebo-treated patients were eligible to enter IM-UNITI but were not included in the primary population (Fig. 1).
In all 3 studies, randomization was performed centrally using permuted blocks. Stratification variables in both induction studies included study region, CDAI score (≤300 or >300), and presence/absence of initial response to TNF-antagonist therapy (only in UNITI-1). In IM-UNITI, stratification variables included ustekinumab induction dose received and remission status at maintenance baseline (week 0).
The independent ethics committee or institutional review board at each study site approved study protocols. All 3 studies were conducted in accordance with the Declaration of Helsinki, Good Clinical Practices, and applicable regulatory requirements. Written informed consent was obtained from all patients prior to enrollment.
2. Study Assessments
The CDAI scores and CRP concentrations were evaluated at weeks 0, 3, 6, and 8 in the induction studies and every 4 weeks in the maintenance study. Fecal calprotectin and lactoferrin concentrations were evaluated at weeks 0 and 6 during induction and at weeks 8, 24, and 44 during maintenance. Safety assessments were performed throughout the study, which included monitoring of treatment-emergent adverse events (TEAEs) and vital signs and clinical laboratory evaluations.
Additionally, serum ustekinumab concentrations were measured using a validated electrochemiluminescent immunoassay method at weeks 0, 3, 6, and 8 during induction and every 4 weeks during maintenance. Antibodies against ustekinumab were evaluated using drug-tolerant electrochemiluminescent assay at weeks 0 and 6 during induction and at weeks 12, 24, 36, and 44 during maintenance.
3. Study Endpoints
1) Induction Studies
The primary endpoint was clinical response (defined as a reduction from baseline in CDAI score ≥100) at week 6. Patients with a baseline CDAI score between 220 and 248 were considered to show a clinical response if a CDAI score of <150 was attained. The major secondary endpoints were clinical remission at week 8 (defined as a CDAI score of <150), clinical response at week 8, and 70-point response (defined as a reduction from baseline in the CDAI score of ≥70) at weeks 3 and 6.
2) Maintenance Study
The primary endpoint was clinical remission at week 44 (defined as a CDAI score of <150). The major secondary endpoints were clinical response at week 44, clinical remission at week 44 among patients in clinical remission to ustekinumab at week 0, corticosteroid-free remission at week 44, and clinical remission at week 44 in the subset of patients who were refractory or intolerant to TNF antagonist therapy (i.e., patients from UNITI-1 induction study).
Other endpoints included change from baseline in CRP, fecal calprotectin, and lactoferrin concentrations during induction and maintenance.
4. Statistical Analyses
Statistical analyses for the overall study population have been described elsewhere.19 In both induction and maintenance studies, comparisons of primary and secondary endpoints between ustekinumab and placebo for the overall study population were performed using Cochran-Mantel-Haenszel chi-square test at a 2-sided significance level of 0.05. To control the overall Type 1 error rate, the primary endpoint and major secondary endpoints were tested in a hierarchical order. Subsequent endpoints were considered statistically significant only if the preceding endpoint in the hierarchy was positive. Patients with treatment failure (i.e., CD-related surgeries, prohibited changes to concomitant CD medications, initiation of prohibited medications), or insufficient data to calculate CDAI scores (i.e., <4 of 8 CDAI components available) were considered unresponsive or not in remission. For maintenance, patients who had a loss of response or discontinued study agent due to lack of therapeutic effect or an adverse event of worsening disease were also considered as treatment failures. Treatment-failure and missing data rules were also applied for dichotomous secondary endpoints. For continuous endpoints, baseline values (from induction week 0) were assigned from time of the treatment failure, and the last available observation was carried forward for missing data. The sample size for Japanese subpopulation was determined with reference to the guidance from Japanese Ministry of Health, Labour and Welfare.2021 The results of the Japanese subpopulation analysis were summarized descriptively, and consistency in efficacy and safety outcomes between the Japanese subpopulation and overall population were evaluated.
RESULTS
1. Patient Disposition
In UNITI-1, a total of 741 patients were randomized, of which 56 were from Japan (placebo, 18; ustekinumab 130 mg, 19; ustekinumab ~6 mg/kg, 19). In UNITI-2, of the 628 patients randomized, 26 were from Japan (placebo, 9; ustekinumab 130 mg, 8; ustekinumab ~6 mg/kg, 9). A total of 397 patients, including 21 patients from Japan, responded to ustekinumab treatment in induction studies and were enrolled in IM-UNITI as the primary (randomized) population (placebo, 4; ustekinumab 90 mg SC q8w group, 9; ustekinumab 90 mg SC q12w group, 8). A total of 884 (Japan 53) patients who were nonresponders to ustekinumab or placebo or who were responders to placebo entered IM-UNITI as a nonrandomized population (Fig. 1). The demographic characteristics and baseline disease characteristics in the Japanese subpopulation were similar to those of the overall study population, except for median body weight, which was lesser, and CRP level, which was greater in the Japanese subpopulation than the overall study population (Table 1).
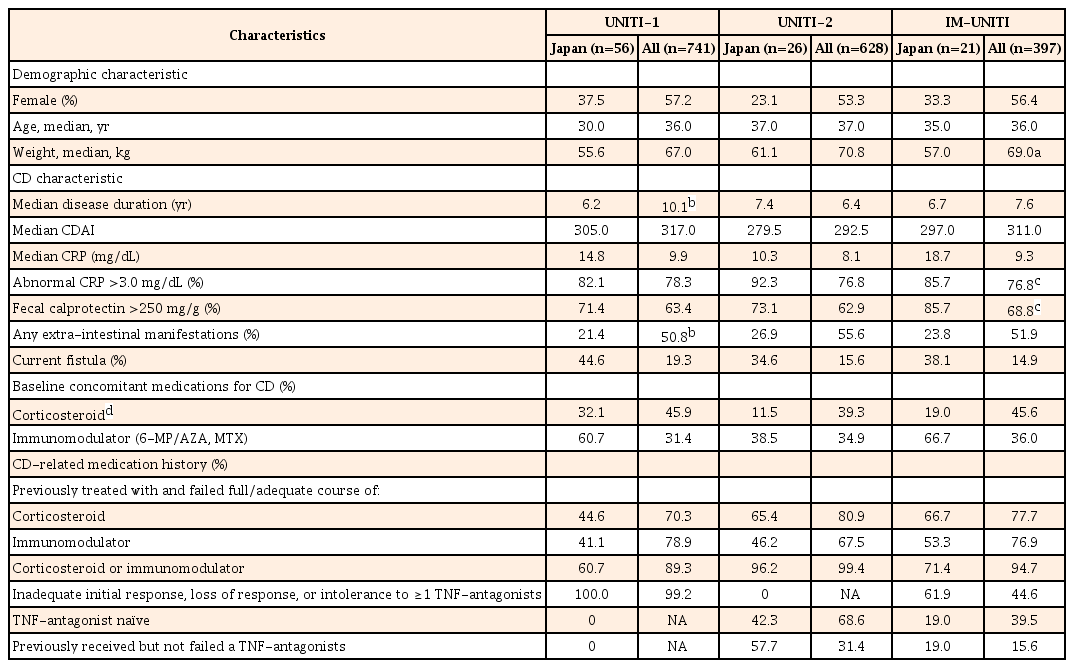
Patient Demographics and Clinical Characteristics of the Overall Study Population and Japanese Subpopulation: Induction Studies and Maintenance Study
2. Efficacy in Induction Studies
1) Primary Efficacy Endpoint
In UNITI-1, the proportion of patients achieving clinical response at week 6 was greater in ustekinumab groups (130 mg: Japanese subgroup, 36.8% [7/19]; overall, 34.3%; ~6 mg/kg: Japanese subgroup, 31.6% [6/19]; overall, 33.7%) than in the placebo group (Japanese subgroup, 21.5% [5/18]; overall, 27.8%) (Fig. 2A). Similarly, in UNITI-2, the proportion of patients achieving clinical response at week 6 was greater in ustekinumab groups (130 mg: Japanese subgroup, 37.5% [3/8]; overall, 51.7%; ~6 mg/kg: Japanese subgroup, 55.6% [5/9]; overall, 55.5%) than in the placebo group (Japanese subgroup, 11.1% [1/9]; overall, 28.7%) (Fig. 2B)
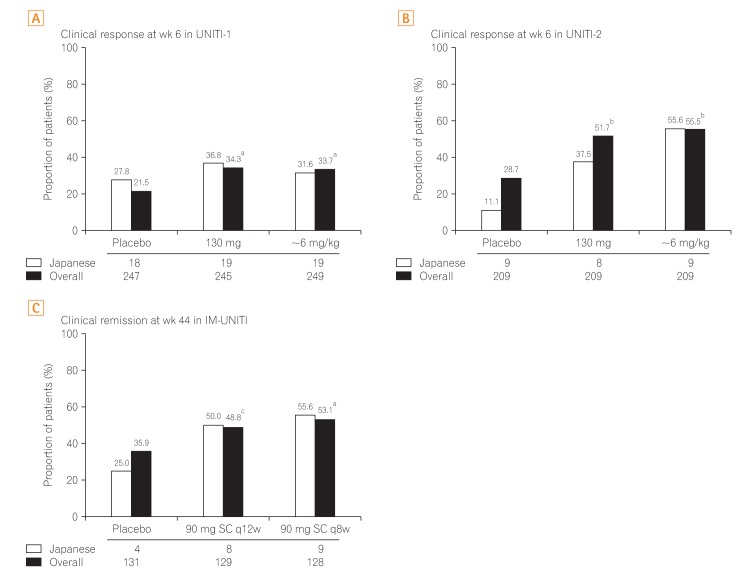
Primary endpoint in the overall study population and Japanese subpopulation. The primary efficacy outcome in induction and maintenance studies observed in Japanese subpopulation were similar to the global study population. aP<0.001 vs. placebo; bP<0.01 vs. placebo; cP=0.04. q8w, every 8 weeks; q12w, every 12 weeks.
2) Major Secondary Efficacy Endpoints
In both the induction studies, a greater proportion of patients treated with ustekinumab (both 130 mg and ~6 mg/kg) achieved clinical response and remission at week 8 than the placebo group (in Japanese subpopulation and overall study population) (Table 2). A higher proportion of patients attained the 70-point response at week 3 or week 6 with either of the ustekinumab induction doses than those in the placebo group, and the response was sustained at week 6 in both the Japanese and overall study populations (Table 2). Thus, the primary and major secondary endpoints were met and both induction doses were better than the placebo in the subset of Japanese patients, and results were generally similar to the overall population.19
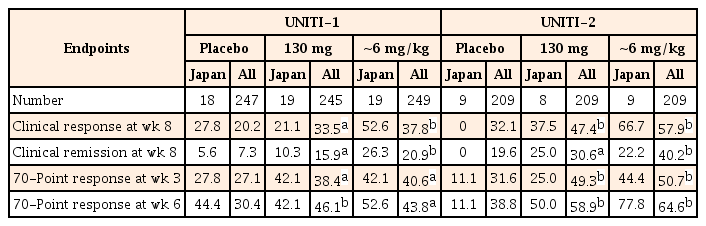
Major Secondary Endpoints of Induction Studies in the Overall Population and Japanese Subpopulation
3) Other Efficacy Endpoints
The median change in CDAI scores in the Japanese subpopulation was generally better in both ustekinumab induction groups when compared with the placebo (Fig. 3A-D). In the Japanese subpopulation, the median serum CRP concentration was considerably reduced at the earliest time-point (week 3) in both the ustekinumab induction groups (130 mg: UNITI-1, −2.3 mg/L; UNITI-2, −1.78 mg/L; ~6 mg/kg: UNITI-1, −3.20 mg/L; UNITI-2, −7.20 mg/L) compared with the corresponding placebo groups (UNITI-1, 2.17 mg/L; UNITI-2, 0.55 mg/L). The reductions seen after ustekinumab treatment were persistent through week 8. These reductions in CRP concentration were also similar to those in the overall population (Supplementary Fig. 1).
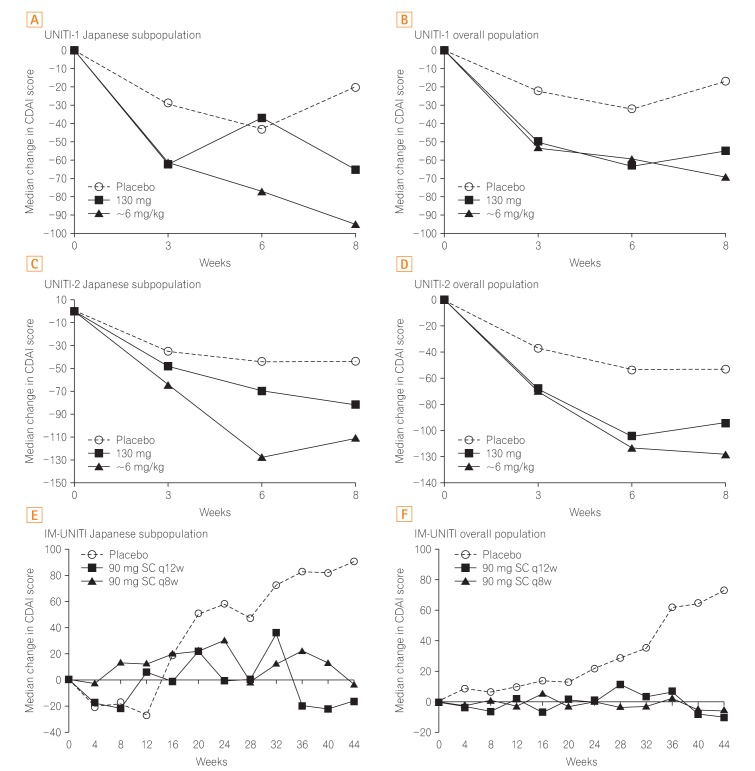
Median change in CDAI scores over time. In the induction studies and maintenance study, the median change in CDAI scores consistently improved with ustekinumab compared with that with placebo in the Japanese subpopulation and overall study population. All P<0.05 vs. placebo in the overall population of the UNITI-1, UNITI-2, and IM-UNITI studies. q12w, every 12 weeks; q8w, every 8 weeks. Panel B, D, and F are adapted from Feagan BG, et al. N Engl J Med 2016;375:1946-1960, with permission Massachusetts Medical Society.19
In the Japanese subpopulation of UNITI-1, the median fecal calprotectin concentration at week 6 was reduced (−6.5 mg/kg) in the ustekinumab 130 mg group, while it was increased by 2.11 mg/kg in the ustekinumab ~6 mg/kg group and by 15.84 mg/kg in the placebo group. In the UNITI-2 Japanese subpopulation, there was no change in the fecal calprotectin concentration at week 6 in the ustekinumab 130 mg group; however, it was reduced (−108.23 mg/kg) in the ustekinumab ~6 mg/kg group and was increased by 2.97 mg/kg in the placebo group. In the overall study population, significantly greater reductions in fecal calprotectin were observed at week 6 in both the ustekinumab induction groups compared with the placebo group (P<0.001) (Supplementary Fig. 2A).
3. Efficacy in Maintenance Study
1) Primary Efficacy Endpoint
The proportion of responders to ustekinumab induction who exhibited clinical remission at week 44 during maintenance was greater in the ustekinumab groups (90 mg SC q12w: overall, 48.8%; Japanese subgroup, 50.0% [4/8]; 90 mg SC q8w: overall, 53.1%; Japanese subgroup, 55.6% [5/9]) than in the placebo group (overall, 35.9%; Japanese subgroup, 25.0% [1/4]) (Fig. 2C).
2) Major Secondary Efficacy Endpoints
Similar to the results observed for the overall study population, a higher proportion of patients in the Japanese subgroup showed sustained clinical remission (remission at weeks 36, 40, and 44) in the ustekinumab groups (90 mg SC: q12W, 37.5% [3/8]; q8w, 55.6% [5/9]) than in the placebo group (25.0% [1/4]) (Supplementary Fig. 3).
The proportion of Japanese patients remaining in remission at week 44 among those in remission after induction, at maintenance baseline (remission in remitters), was 80% (4/5) and 50% (2/4) in the ustekinumab 90 mg SC q8w and q12w groups, respectively, while 1/1 patient maintained remission on SC placebo (i.e., the only patient in remission, when randomized to SC placebo maintenance, stayed in remission at week 44). In the overall study population, a greater proportion of patients who were in remission at maintenance baseline maintained remission with ustekinumab 90 mg SC q8w treatment (67%) than those with q12w (56%) and placebo (46%) treatments. Number of patients in the Japanese subgroup achieving corticosteroid-free remission at week 44 were greater with ustekinumab maintenance (90 mg SC: q12w, 55.6% [5/9]; q8w, 50% [4/8]) than with placebo (25% [1/4]). In the overall study population, the proportion of patients in corticosteroid-free remission at week 44 was significantly greater in the q8w group (P=0.004) and nominally significant in the q12w group (P=0.035; as per hierarchical testing procedure) than with placebo (Supplementary Fig. 4).
In the Japanese subpopulation, clinical remission at week 44 among those who were refractory or intolerant to TNF antagonist therapy (subset of patients from UNITI-1) was achieved with 90 mg SC q12w (33.3% [2/6]) and q8w ustekinumab treatment (50.0% [2/4]), but not with placebo treatment (0% [0/3]). Similarly, in the subset of UNITI-1 patients from the overall population, a greater proportion of patients receiving ustekinumab 90 mg SC q12w (38.6%) and q8w (41.1%) maintained clinical remission at week 44 than those on placebo (26.2%) (Supplementary Fig. 5).
3) Other Efficacy Endpoints
Throughout the maintenance study, the median change in CDAI scores was generally improved in both the ustekinumab dose groups and was better than in the placebo group for both the Japanese subpopulation and the overall study population (Fig. 3 E and F).
A normal CRP level at week 44 was achieved in Japanese patients who had baseline CRP >3 mg/L in the induction studies, when treated with ustekinumab 90 mg SC q12w (42.9% [3/7]), while normal levels were not achieved with 90 mg SC q8w (0/8 patients) or placebo (0/3 patients) treatment. However, in the overall population, proportion of patients attaining a normalized CRP was greater for both the ustekinumab regimens than the placebo. The median change in fecal calprotectin concentration at week 44 in the Japanese subpopulation was considerably reduced with both 90 mg SC q12w (−102.3 mg/kg) and q8w treatment (−17.90 mg/kg), while it increased over the maintenance period in the SC placebo group (168.15 mg/kg). In the overall study population, there was no change in fecal calprotectin levels at week 44 in both the ustekinumab groups, while it increased in the placebo group (153.85 mg/kg) (Supplementary Fig. 2B).
In the Japanese subpopulation, when nonrandomized patients (nonresponders to ustekinumab IV induction, n=31; Fig. 1) of the maintenance study were treated with an additional SC dose of ustekinumab (90 mg) at week 0 of maintenance, 61.3% (n=19) achieved clinical response 8 weeks later. A total of 22 of these nonrandomized patients continued ustekinumab, receiving 90 mg q8w maintenance regimen. Of these patients, 54.5% (n=12) maintained clinical response and 36.4% (n=8) achieved clinical remission at week 44. In the overall study population, 50.5% of nonrandomized patients achieved clinical response at week 8 (after ustekinumab 90 mg SC injection at week 0) and 68.1% of those patients continued to maintain a clinical response and 50.2% were in remission at week 44.
4. Safety
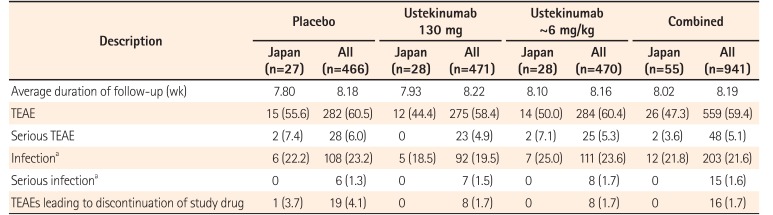
In the combined Japanese subpopulations of both induction studies, TEAEs were reported in 44.4% of patients receiving ustekinumab (130 mg), 50.0% in the ~6 mg/kg group, and 55.6% in the placebo group. A consistent pattern was observed in the overall study population (ustekinumab 130 mg, 58.4%; ~6 mg, 60.4%; placebo, 60.5%) (Table 3). The most frequently reported TEAEs (>2 patients in ustekinumab groups) in the Japanese subpopulation of induction studies were nasopharyngitis (n=6, 10.9%) and pyrexia (n=3, 5.5%). Similarly, in the overall population, TEAEs of nasopharyngitis (4.6%) and pyrexia (5.9%) were commonly reported. The incidence of infection TEAEs was similar across the treatment groups in Japanese patients (ustekinumab 130 mg, 18.5%; ~6 mg/kg, 25.0%; placebo, 22.2%) and none of these were serious infections. The proportion of overall serious TEAEs in the induction studies was also not higher following ustekinumab treatment and occurred at similar frequencies in the Japanese subpopulation (both ustekinumab doses, 3.6%; placebo, 7.4%) and the overall study population (both ustekinumab doses, 5.1%; placebo, 6.0%). One patient in the placebo group experienced a TEAE leading to study drug discontinuation (Table 3).
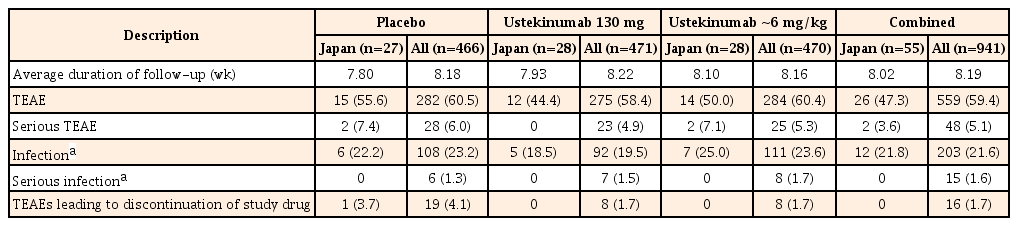
Summary of Safety Results through Week 8 (Combined UNITI-1 and-2 Study)
In the IM-UNITI maintenance study, TEAEs occurred at similar frequencies across the treatment groups in both the Japanese subpopulation and the overall study population (Table 4). The most frequently reported TEAEs (>2 patients in ustekinumab groups) in the Japanese subpopulation were nasopharyngitis (n=6, 35.3%) and headache (n=4, 23.5%). The other infections observed in ≥2 Japanese patients in maintenance were anal abscess, pharyngitis, and vulvovaginal candidiasis (n=2, 11.8%; each), and this was similar to the values for the overall study populations. A total of 3 serious TEAEs (q12w, n=2; q8w, n=1) were observed in the ustekinumab group within the Japanese subpopulation. Two of these were serious infections (anal abscess and bacteremia) observed in the 90 mg SC q12w group. The serious TEAE of anal abscess was the only adverse event leading to discontinuation of the study agent (Table 4).
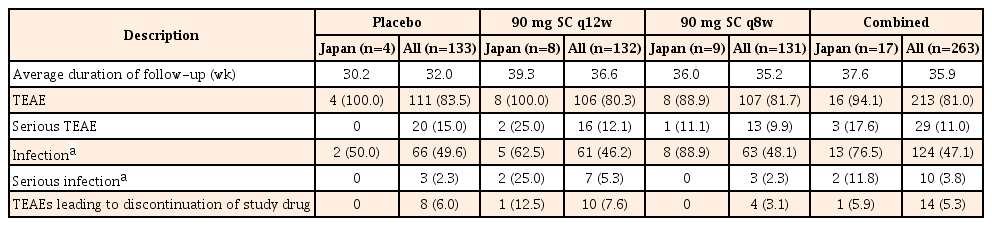
Summary of Safety Results through Week 44 of the IM-UNITI Study
There were no deaths, malignancies, events of tuberculosis, or opportunistic infections reported in the Japanese subpopulation through 52 weeks of therapy. None of the Japanese patients in the ustekinumab group reported any TEAEs related to infusions in the induction or maintenance studies.
5. Pharmacokinetics and Immunogenicity
No clear difference in serum ustekinumab concentrations was observed between Japanese patients and non-Japanese patients in the induction studies or the maintenance study, regardless of the dose received (Supplementary Figs 6 and 7). In the overall population, 2 patients who received ustekinumab (130 mg) IV developed antibodies to ustekinumab in the induction studies and 27 patients (2.3%) developed antibodies in the maintenance study, determined using a drug tolerant assay. None of the Japanese patients who received ustekinumab developed antibodies to ustekinumab in the induction studies and 2 Japanese patients (9.5%) developed antibodies in the maintenance study.
DISCUSSION
Genetic variations that may increase predisposition to CD or affect treatment responses among ethnic populations have been reported. For example, there are certain coding variants in nucleotide-binding oligomerization domain-containing protein 2 (NOD2) that have a large effect on IBD risk in Caucasians; however, such variants in allele frequency in Asian populations, including Japanese population, is absent, which suggests that there is genetic heterogeneity in predisposition to CD.2223 Furthermore, the treatment effect of thiopurine may differ between Asians and Caucasians because of the prevalence of a variant in NUDT15, which is associated with thiopurine-induced leukopenia in patients with IBD.24 Because differences in IBD treatment responses among ethnic populations are possible due to genetic heterogeneity,24 it is important to assess the consistency or difference of treatment outcomes between specific ethnic populations and overall population.
The phase 3 global clinical study program of ustekinumab studied efficacy and safety in a wide range of patients with moderately to severely active CD who had been previously treated with TNF-antagonists or conventional therapy.19 In the Japanese subpopulation of this phase 3 program, ustekinumab 130 mg and ~6 mg/kg IV induction therapy showed increased clinical response rates at week 6 over placebo, both among patients who were refractory to TNF-antagonists as well as to conventional therapy. Among patients who responded to IV ustekinumab in the induction studies, treatment with SC ustekinumab (90 mg q12w and q8w) maintenance therapy demonstrated increased rates of clinical remission and response at week 44.
The primary and major secondary efficacy outcomes in induction and maintenance studies observed in the Japanese subpopulation were consistent with the findings observed in the overall global study population.19 In both induction studies, ~6 mg/kg ustekinumab showed higher rates of clinical response and remission than 130 mg dose, particularly at week 8. Patients in the UNITI-2 study who had failed responses only to conventional therapy achieved higher rates of response and remission than the patients with failed responses to TNF-antagonist in the UNITI-1 study, possibly because these patients had a less refractory disease. Similar to the overall study population, in the Japanese subpopulation treated with ustekinumab, induction demonstrated greater reductions in CRP concentration than the placebo even at the first post-baseline visit at week 3. Reductions in fecal calprotectin levels were observed only with 130 mg ustekinumab treatment in UNITI-1 study and ~6 mg/kg ustekinumab treatment in UNITI-2 study in the Japanese subpopulation; whereas in the overall study population, both doses of ustekinumab showed reductions in fecal calprotectin and ~6 mg ustekinumab dose showed the highest reduction in both studies. This may be due to high variability in calprotectin levels and limited number of Japanese patients. However, overall treatment with ustekinumab improved objective markers of inflammation along with improvements in clinical response and remission in the Japanese subpopulation similar to the overall study population.
In the maintenance study, in Japanese patients who responded to IV ustekinumab induction, treatment with SC ustekinumab (90 mg q12w and q8w) maintained clinical remission, clinical response, remission among those in remission after induction, and corticosteroid-free remission as well as sustained remission. Although both doses of SC ustekinumab (90 mg q12w and q8w) demonstrated improvement in clinical outcomes, q8w dose showed the highest improvement in both the Japanese and overall populations. In Japanese patients who had elevated CRP concentration (>3 mg/L) at baseline of induction studies, treatment with ustekinumab 90 mg SC q12w normalized CRP levels at week 44. A considerable reduction in fecal calprotectin levels at week 44 was also observed with ustekinumab maintenance regimens in the Japanese subpopulation. Japanese patients who were nonresponders to IV ustekinumab during induction (nonrandomized patients) also showed greater response during the additional maintenance ustekinumab SC treatment, which corroborated the results of the overall study population as well as results in the randomized primary population.
Ustekinumab IV induction and SC maintenance regimens were well tolerated in the Japanese subpopulation and in the overall study population. The rates of TEAEs and serious TEAEs were similar across the treatment groups. There were no reports of serious infections with ustekinumab IV treatment during induction in the Japanese subpopulation; whereas only 2 patients reported serious infections with ustekinumab 90 mg SC q12w during maintenance. Treatment with ustekinumab was associated with only one TEAE leading to treatment discontinuation. Additionally, there were no reports of infusion reactions, and no deaths were reported in these studies through week 44, in either the Japanese or overall populations. Overall, ustekinumab treatment showed no safety concerns and results were consistent with those for the overall study population as well as for those with the previous studies conducted in CD patients1819 and with previous long-term studies conducted for other indications, including psoriasis and psoriatic arthritis.17252627 No clear difference in serum ustekinumab concentrations was observed between the Japanese patients and non-Japanese patients in induction studies or the maintenance study.
The sample size in this Japanese subpopulation analysis was small to perform statistical analysis; however, efficacy and safety outcomes in Japanese patients were analogous to the results of global population.
To the best of our knowledge, this is the first global IBD study to demonstrate findings of the Japanese subpopulation consistent to that of the overall study population. The IV ustekinumab as an induction therapy was more effective than placebo in Japanese patients with moderate to severely active CD, who were refractory to either TNF antagonists or conventional therapy. Among the responders to IV ustekinumab induction, treatment with SC ustekinumab was also more effective than placebo in maintaining clinical response and remission along with other efficacy outcomes. Both ustekinumab induction and maintenance regimens were generally well tolerated. Despite genetic variations that may exist in the Japanese population, these data suggest that ustekinumab provides efficacy in Japanese patients with CD, similar to observations in the global population. Ustekinumab treatment exhibits a favorable benefit-risk balance for CD; therefore, it could be considered as a new therapeutic option for moderate to severely active CD in Japanese patients who have failed conventional therapies or have additionally previously failed TNF antagonists.
ACKNOWLEDGEMENTS
The authors thank the study participants without whom this study would never have been accomplished and also thank the investigators for their participation in the study. The authors also thank Dr. Takayuki Ota for his contribution to this manuscript. Writing assistance was provided by Ramji Narayanan, ISMPP CMPPTM (SIRO Clinpharm Pvt. Ltd., Thane, India), funded by Janssen Pharmaceutical K.K., Japan and publication support was provided by Kenichiro Tsutsumi (Janssen Pharmaceutical K.K., Tokyo, Japan). This study was funded by Janssen Research & Development, LLC and Janssen Pharmaceutical K.K., Japan.
Notes
AUTHOR CONTRIBUTIONS: T.H. and C.S. were involved in conception and design of the study. T.H. was also involved in acquisition of data. R.Z. was involved in analysis of data. All authors were involved in interpretation of data, drafting and critical revision of the manuscript and have approved the final manuscript for submission. All authors also had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Financial support: This study was funded by Janssen Research & Development, LLC and Janssen Pharmaceutical K.K., Japan.
Conflict of interest: Yuya Imai, Yoko Murata, Nobuko Matsushima, and Richuan Zheng are employees of Janssen Pharmaceutical K.K., Japan. Dr. Christopher Gasink is an employee of Janssen Research & Development, LLC, United States. Dr. Toshifumi Hibi is a consultant for Janssen Pharmaceutical K.K., Japan.
This paper was presented at 7th Annual Meeting of Japanese Society of Inflammatory Bowel Disease (JBSID) 2016, July 9-10, Kyoto, Japan.
References
SUPPLEMENTARY MATERIALS
Supplementary Fig. 1
Change in CRP concentration through week 8 of induction studies. Panel B and D are adapted from Feagan BG, et al. N Engl J Med 2016;375:1946-1960, with permission Massachusetts Medical Society.19
Supplementary Fig. 2
Change in fecal calprotectin concentration at: (A) week 6 of the induction studies; (B) week 44 of the maintenance study. aP<0.001 vs. placebo; bP=0.002 vs. placebo. q12w, every 12 weeks; q8w, every 8 weeks.
Supplementary Fig. 3
Sustained clinical remission at weeks 36, 40, and 44 of the maintenance study. aP=0.023 vs. placebo; bP<0.001 vs. placebo. q12w, every 12 weeks; q8w, every 8 weeks.
Supplementary Fig. 4
Corticosteroid-free remission at week 44 of the maintenance study. aP=0.035 vs. placebo but only nominally significant per the hierarchical testing procedure; bP=0.004 vs. placebo. q12w, every 12 weeks; q8w, every 8 weeks.
Supplementary Fig. 5
Clinical remission at week 44 in patients who were refractory or intolerant to tumor necrosis factor-antagonist therapy (UNITI-1 patients). q12w, every 12 weeks; q8w, every 8 weeks.
Supplementary Fig. 6
Mean±SD serum ustekinumab concentrations (µg/mL) through week 8 (A) in UNITI-1 and (B) in UNITI-2 studies.
Supplementary Fig. 7
Mean±SD serum ustekinumab concentrations (µg/mL) in IM-UNITI (A) in the 90 mg SC q12w group and (B) in the 90 mg SC q8w group. q12w, every 12 weeks; q8w, every 8 weeks.