Efficacy and safety of filgotinib as induction and maintenance therapy for Japanese patients with moderately to severely active ulcerative colitis: a post-hoc analysis of the phase 2b/3 SELECTION trial
Article information

Abstract
Background/Aims
The safety and efficacy of filgotinib, a once-daily oral Janus kinase 1 preferential inhibitor, were evaluated in Japanese patients with ulcerative colitis (UC) in the phase 2b/3 SELECTION trial.
Methods
SELECTION (NCT02914522) was a randomized, placebo-controlled trial comprising 2 induction studies and a maintenance study. Adults with moderately to severely active UC were randomized in induction study A (biologic-naïve) or B (biologic-experienced) to receive filgotinib 200 mg, 100 mg, or placebo once daily for 11 weeks. Patients in clinical remission or Mayo Clinic score response at week 10 entered the 47-week maintenance study. Efficacy and safety outcomes were assessed in Japanese patients enrolled in Japan.
Results
Overall, 37 and 72 Japanese patients were enrolled in Japan in induction studies A and B, respectively, and 54 entered the maintenance study. Numerically higher proportions of filgotinib 200 mg-treated than placebo-treated patients achieved clinical remission in induction study A (4/15 [26.7%] vs. 0/6 [0%]) and the maintenance study (5/20 [25.0%] vs. 0/9 [0%]), but not induction study B (1/29 [3.4%] vs. 1/14 [7.1%]). Both doses were well tolerated, and no new safety signals were noted. Herpes zoster was reported in 1 filgotinib 200 mg-treated patient in each of induction study A (2.3%, 1/44) and the maintenance study (5.0%, 1/20).
Conclusions
These data, alongside those of the overall SELECTION population, suggest the potential of filgotinib 200 mg as a viable treatment option for Japanese patients with UC. Owing to small patient numbers, data should be interpreted cautiously.
INTRODUCTION
Ulcerative colitis (UC) is a chronic and debilitating inflammatory bowel disease leading to symptoms such as rectal bleeding, frequent bowel movements, and tenesmus [1,2]. In addition, many patients with UC experience anemia, fatigue, and reduced health-related quality of life [3]. In cases of severe or treatment-refractory disease, surgery may be required [2]. Although the incidence of UC is stabilizing in Western countries [4], prevalence of the disease has risen substantially in Japan over the past 2 decades [5]. In Japan, UC is designated as an intractable disease by the Ministry of Health, Labour and Welfare [6]. Approximately 220,000 Japanese patients with UC were recorded in a nationwide survey in Japan in 2015 [5,7].
UC treatment goals in Japan align with those in many other countries and include treat-to-target aims of achieving absence of rectal bleeding, reduced stool frequency, and a Mayo endoscopic subscore (MES) of 0 or 1, with histologic remission as an adjunct goal [8,9]. Currently available treatment options in Japan for moderately to severely active UC include 5-aminosalicylate, corticosteroids, immunosuppressants (such as calcineurin inhibitors), immunomodulators (such as azathioprine), leukocyte apheresis (particularly in corticosteroid-refractory or corticosteroid-dependent patients [10]), biologic therapies (infliximab, adalimumab, golimumab, vedolizumab, and ustekinumab) and the Janus kinase (JAK) inhibitor (tofacitinib).
Choice of biologic therapies for UC was dominated by tumor necrosis factor (TNF) antagonists for nearly 20 years, but anti-integrin and anti-interleukin-12/interleukin-23 antibodies have recently become available [11,12]. Although biologic therapies have led to substantial improvements in the care of patients with UC [1], not all patients experience benefits [13] and many patients lose response to treatment over time and need to switch therapies [14]. Currently, the only JAK inhibitor approved for the treatment of UC in Japan is tofacitinib. Consequently, more treatment options are needed for patients with UC.
Filgotinib is a once-daily, oral JAK1 preferential inhibitor. JAK inhibitors block one or more JAKs to inhibit the JAK–signal transducer and activator of transcription pathway and oppose the biological effects of inflammatory cytokines involved in the pathogenesis of UC [15]. Filgotinib preferentially inhibits JAK1 over JAK2, JAK3, and TYK2 [16], which may confer an improved safety profile [17-19]. Filgotinib is approved in Europe and Japan for the treatment of adults with moderately to severely active rheumatoid arthritis [20,21]. In a phase 1 study in healthy volunteers, the pharmacokinetic profiles of filgotinib and its active metabolite (GS-829845) were shown to be similar in Japanese and Caucasian participants [22].
The international phase 2b/3 SELECTION trial (ClinicalTrials.gov ID: NCT02914522) evaluated the efficacy and safety of filgotinib in patients with moderately to severely active UC [23]. Filgotinib 200 mg was efficacious in inducing clinical remission among biologic-naïve and biologic-experienced patients compared with placebo. Furthermore, a greater proportion of patients treated with filgotinib 200 mg than with placebo during the maintenance phase achieved clinical remission, sustained clinical remission, and 6-month corticosteroid-free remission by week 58. Both filgotinib 200 mg and 100 mg were well tolerated. These post-hoc analyses aim to evaluate the safety and efficacy of filgotinib induction and maintenance therapy in Japanese patients enrolled in Japan with moderately to severely active UC who were included in the SELECTION trial.
METHODS
1. Study Design
SELECTION was a phase 2b/3 randomized, placebo-controlled trial comprised of 2 induction studies and a maintenance study. The study design is shown in Supplementary Fig. 1. Eligible patients were enrolled into 1 of 2 induction studies: induction study A (biologic-naïve patients) or induction study B (biologic-experienced patients). Patients were randomized 2:2:1 to filgotinib 200 mg, filgotinib 100 mg, or placebo orally once daily for 11 weeks. Randomization was performed using an interactive web response system. In induction study A, patients were stratified by use of oral systemic corticosteroids on day 1 and use of immunosuppressants (6-mercaptopurine, azathioprine, and methotrexate) on day 1. In induction study B, patients were stratified by the same factors as induction study A and by previous exposure to one versus more than one biologic agent. In the maintenance study, patients were stratified by the same factors as induction study A and by participation in induction study A or B.
Patients who achieved clinical remission (MES of 0 or 1, rectal bleeding subscore of 0, and a ≥ 1-point decrease in stool frequency from induction baseline to achieve a subscore of 0 or 1) or a Mayo Clinic score (MCS) response (reduction of ≥ 3 points in MCS and ≥ 30% from induction baseline with an accompanying decrease in rectal bleeding subscore of ≥ 1 point, or an absolute rectal bleeding subscore of 0 or 1) at week 10 were considered responders and could enter the 47-week maintenance study. Responders who received induction filgotinib were re-randomized 2:1 to receive their induction filgotinib dose or placebo. Placebo responders continued to receive placebo. Full details of the study design have been reported by Feagan et al. [23]. The study was conducted in accordance with the International Conference on Harmonisation of Technical Requirements For Registration Of Pharmaceuticals For Human Use Guideline For Good Clinical Practice and the Declaration of Helsinki. The trial protocol, protocol amendments, consent forms, patient information sheets, administrative letters, and advertisements were submitted by each investigator to each independent ethics committee or institutional review board (Kitasato University Shirokane Institutional Review Board and others) for review, and approved before trial initiation. All patients provided written informed consent.
2. Participants
Eligible patients were 18–75 years old at screening and had a diagnosis of UC with endoscopic and histopathologic evidence of UC for at least 6 months before enrollment. Patients had moderately to severely active UC (MES ≥ 2, rectal bleeding subscore ≥ 1, stool frequency subscore ≥ 1, physician’s global assessment subscore ≥ 2; and a total MCS of 6–12). Biologic-naïve patients entered induction study A; biologic-experienced patients who had previously experienced an inadequate clinical response, loss of response, or intolerance to any TNF antagonist, vedolizumab, or any other biologic agent, entered induction study B. Patients who had previously received a JAK inhibitor were not permitted to enter either induction study, following an amendment to the protocol. Patients who underwent leukocyte apheresis within 2 months before screening were also excluded from the study. Further details of patient inclusion and exclusion criteria have been described by Feagan et al. [23]. Stable doses of concomitant corticosteroids were permitted up to week 14, but had to be tapered according to a predefined schedule thereafter [23].
3. Outcome Measures and Assessments
Described below are the efficacy and safety outcomes of the SELECTION trial. In these post-hoc analyses, we assessed these outcomes in Japanese patients who participated in the trial in Japan. The primary outcomes of the SELECTION trial were the proportions of patients in clinical remission at week 10 in induction studies A and B, and at week 58 in the maintenance study. Clinical remission was assessed using symptoms of rectal bleeding and stool frequency recorded daily by patients in an eDiary as well as endoscopic and histopathologic data from centrally read colonoscopies/flexible sigmoidoscopies, with biopsies performed at baseline, week 10, and week 58.
Key secondary outcomes were the proportions of patients in MCS remission (total MCS of ≤ 2 and no single subscore > 1), MCS remission alternative definition (rectal bleeding, stool frequency, and physician’s global assessment subscores of 0; an endoscopic subscore of 0 or 1; and an overall MCS of 1 or 0), endoscopic remission (MES of 0), and histologic remission (grade 0 Geboes score of ≤ 0.3, grade 1 score of ≤ 1.1, grade 2A score of ≤ 2A.3, grade 2B score of 2B.0, grade 3 score of 3.0, grade 4 score of 4.0, and grade 5 score of 5.0) at week 10 in induction studies A and B, and at week 58 in the maintenance study. Further key secondary outcomes were the proportions of patients in 6-month corticosteroid-free clinical remission (clinical remission with no corticosteroid use for the indication of UC for ≥ 6 months before week 58 among patients who were receiving corticosteroids at the baseline of the maintenance study) and sustained clinical remission (clinical remission at both week 10 and week 58) at week 58 in the maintenance study.
Exploratory efficacy outcomes included the proportions of patients who achieved an MCS response (reduction of ≥ 3 points in MCS and ≥ 30% from induction baseline with an accompanying decrease in rectal bleeding subscore of ≥ 1 point, or an absolute rectal bleeding subscore of 0 or 1) at week 10 in induction studies A and B, and at week 58 in the maintenance study; change from baseline in partial MCS (pMCS; the sum of rectal bleeding, stool frequency, and physician’s global assessment subscores) over time during the induction and maintenance studies; and endoscopic response (MES of 0 or 1) at week 10 in induction studies A and B, and at week 58 in the maintenance study.
Symptomatic remission (Mayo rectal bleeding subscore of 0, and a ≥ 1-point decrease in stool frequency subscore from induction baseline to achieve a subscore of 0 or 1) at week 10 in induction studies A and B, and at week 58 in the maintenance study, was also evaluated as a post-hoc analysis.
Safety outcomes comprised adverse events (AEs; including AEs of interest), concomitant medications, laboratory analyses, vital signs, electrocardiograms, and physical examinations. AEs of interest included all infections, serious infections, herpes zoster infections, opportunistic infections, malignancies, gastrointestinal perforations, and thromboembolic events (venous thrombosis, pulmonary embolism, arterial thrombosis, and cerebrovascular events). The severity of AEs and clinical laboratory results was graded using the modified Common Terminology Criteria for Adverse Events, Version 4.03.
4. Statistical Analyses
In these post-hoc analyses, efficacy outcomes for induction studies A and B were analyzed for all randomized Japanese patients enrolled in Japan who received at least 1 dose of study drug during the study. For the maintenance study, efficacy outcomes were analyzed for all Japanese patients enrolled in Japan who received filgotinib during induction, were re-randomized, and received at least 1 dose of study drug in the maintenance study. Safety outcomes were analyzed using data from all Japanese patients enrolled in Japan who took at least 1 dose of study drug within each study. Safety analyses for the induction studies were conducted for patients in induction studies A and B combined. Given that these were post-hoc analyses, efficacy outcomes, along with baseline demographics, characteristics, and safety data, were analyzed using descriptive statistics. Missing data for efficacy outcomes were handled using a non-responder imputation approach, with the exception of change from baseline in pMCS, for which a last observation carried forward approach was used. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
RESULTS
1. Patient Disposition and Baseline Characteristics
Overall, 37 biologic-naïve Japanese patients were randomized and received at least 1 dose of study drug in induction study A, and 35 patients (94.6%) completed dosing through to week 10; 72 biologic-experienced Japanese patients were randomized and received at least 1 dose of study drug in induction study B, and 67 patients (93.1%) completed dosing through to week 10 (Supplementary Fig. 2). Overall, 102 Japanese patients completed the induction studies, and of those, 54 achieved clinical remission or an MCS response and entered the maintenance study (Supplementary Fig. 3). Two patients (5.4%) in induction study A (1 patient in each of the filgotinib 100 mg and placebo groups) and 5 patients (6.9%) in induction study B (1 patient in each of the filgotinib 200 mg and 100 mg groups, and 3 patients in the placebo group) prematurely discontinued treatment with the study drug. The most common reason for discontinuation of the study drug was AEs.
In the maintenance study, 5 patients (25.0%) in the filgotinib 200 mg group and 8 patients (88.9%) in the respective placebo group, 6 patients (42.9%) in the filgotinib 100 mg group and 3 patients (50.0%) in the respective placebo group, and 3 patients (60.0%) in the induction placebo/maintenance placebo group prematurely discontinued the study drug (Supplementary Fig. 3). The most common reasons for discontinuation of the study drug were protocol-specified disease worsening and AEs.
Baseline characteristics of Japanese patients in induction studies A and B (Table 1) were largely similar to those of the overall SELECTION population with the exception of mean body mass index, which was lower in the Japanese population [23]. In addition, numerically higher proportions of Japanese patients had an endoscopic subscore of 3 at baseline (biologic-naïve, 64.9%; biologic-experienced, 84.7%) than those in the overall population (biologic-naïve, 55.8%; biologic-experienced, 77.8%). A substantial proportion of biologic-experienced Japanese patients (87.5%) had experienced failure of a TNF antagonist. A numerically lower proportion of biologic-experienced Japanese patients (16.7%) had used vedolizumab compared with biologic-experienced patients in the overall population (57.2%) [23]. Baseline characteristics of Japanese patients in the maintenance study (Supplementary Table 1) were generally similar to those of the overall population [23].
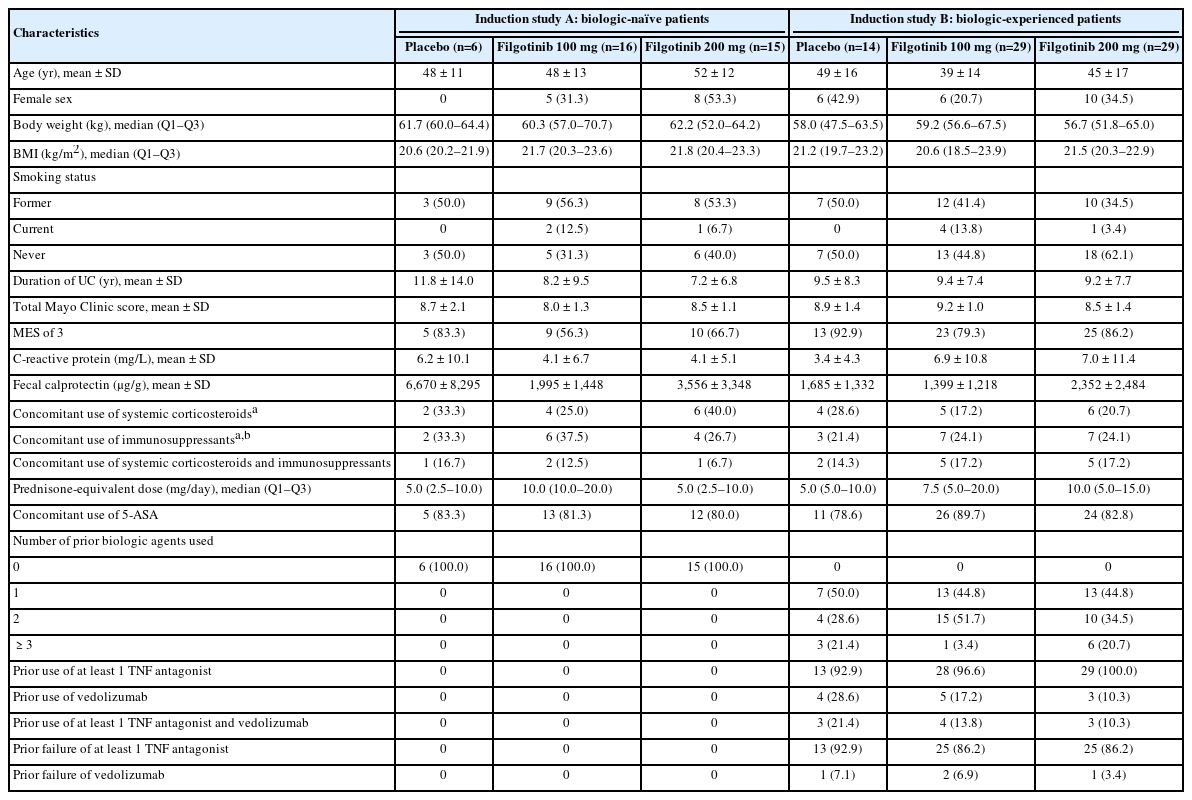
Baseline Demographics and Characteristics of Japanese Patients in Induction Study A and Induction Study B
2. Efficacy
1) Primary Endpoint in SELECTION
A numerically higher proportion of biologic-naïve Japanese patients who received filgotinib 200 mg achieved clinical remission at week 10 than those who received placebo (26.7% [4/15] vs. 0% [0/6], Δ 26.7%, 95% confidence interval [CI], −7.4% to 60.7%) (Fig. 1A). The proportion of biologic-naïve Japanese patients who achieved clinical remission at week 10 was numerically lower in the filgotinib 100 mg group (6.3% [1/16]) than in the filgotinib 200 mg group. The proportions of biologic-experienced Japanese patients who achieved clinical remission at week 10 were low across treatment groups (filgotinib 200 mg, 3.4% [1/29]; filgotinib 100 mg, 6.9% [2/29]; placebo, 7.1% [1/14]) (Fig. 1B).

Proportion of Japanese patients achieving clinical remission (A, B), MCS remission (C, D), MCS remission (alternative definition) (E, F), endoscopic remission (G, H), and histologic remission (I, J) in induction studies A and B at week 10. Error bars indicate 95% confidence intervals (CIs). CIs for 0-responder treatment arms are not presented. CIs for treatment differences between 0-responder treatment arms are not presented. 95% CIs were calculated based on normal approximation with a continuity correction. Clinical remission was defined as an MES of 0 or 1, rectal bleeding subscore of 0, and at least a 1-point decrease in stool frequency from induction baseline to achieve a subscore of 0 or 1. MCS remission was defined as a total MCS of not more than 2 and no single subscore greater than 1. MCS remission (alternative definition) was defined as rectal bleeding, stool frequency, and physician’s global assessment subscores of 0; an endoscopic subscore of 0 or 1; and an overall MCS of 1 or 0. Endoscopic remission was defined as an MES of 0. Histologic remission was defined as grade 0 Geboes score of ≤0.3, grade 1 score of ≤1.1, grade 2A score of ≤2A.3, grade 2B score of 2B.0, grade 3 score of 3.0, grade 4 score of 4.0, and grade 5 score of 5.0. MCS, Mayo Clinic score; MES, Mayo endoscopic subscore.
In the maintenance study, a numerically higher proportion of Japanese patients in the filgotinib 200 mg group achieved clinical remission at week 58 than in the respective placebo group (25.0% [5/20] vs. 0% [0/9], Δ 25.0%, 95% CI, −2.0% to 52.0%) (Fig. 2A). Comparable proportions of biologic-naïve and -experienced Japanese patients treated with filgotinib 200 mg achieved clinical remission at week 58 versus placebo (biologic-naïve, 28.6% [2/7] vs. 0% [0/4]; biologic-experienced, 23.1% [3/13] vs. 0% [0/5]). Numerically higher proportions of Japanese patients in the maintenance filgotinib 100 mg group achieved clinical remission than in the respective placebo group (biologic-naïve, 14.3% [1/7] vs. 0% [0/2]; biologic-experienced, 28.6% [2/7] vs. 25.0% [1/4]) (Supplementary Fig. 4A and B).

Proportion of Japanese patients achieving clinical remission (A), 6-month corticosteroid-free clinical remission (B), sustained clinical remission (C), MCS remission (D), MCS remission (alternative definition) (E), endoscopic remission (F), and histologic remission (G) in the maintenance study at week 58. Separate comparisons were conducted between the filgotinib 200 mg group and the respective placebo group, and the filgotinib 100 mg group and the respective placebo group in the maintenance study. Error bars indicate 95% confidence intervals (CIs). CIs for 0-responder treatment arms are not presented. CIs for treatment differences between 0-responder treatment arms are not presented. 95% CIs were calculated based on normal approximation with a continuity correction. Clinical remission was defined as an MES of 0 or 1, rectal bleeding subscore of 0, and at least a 1-point decrease in stool frequency from induction baseline to achieve a subscore of 0 or 1. Six-month corticosteroid-free clinical remission was defined as clinical remission with no corticosteroid use for the indication of UC for at least 6 months before week 58 among patients who were receiving corticosteroids at the baseline of the maintenance study. Sustained clinical remission was defined as clinical remission at both week 10 and week 58. MCS remission was defined as a total MCS of not more than 2 and no single subscore greater than 1. MCS remission (alternative definition) was defined as rectal bleeding, stool frequency, and physician’s global assessment subscores of 0; an endoscopic subscore of 0 or 1; and an overall MCS of 1 or 0. Endoscopic remission was defined as an MES of 0. Histologic remission was defined as grade 0 Geboes score of ≤0.3, grade 1 score of ≤1.1, grade 2A score of ≤2A.3, grade 2B score of 2B.0, grade 3 score of 3.0, grade 4 score of 4.0, and grade 5 score of 5.0. aOnly patients who received corticosteroids before enrollment were included. MCS, Mayo Clinic score; MES, Mayo endoscopic subscore; UC, ulcerative colitis.
2) Key Secondary Endpoints in SELECTION
A numerically higher proportion of biologic-naïve Japanese patients who received filgotinib 200 mg achieved MCS remission, MCS remission (alternative definition), endoscopic remission, and histologic remission at week 10 than patients who received placebo (Fig. 1). A numerically higher proportion of biologic-naïve Japanese patients treated with filgotinib 100 mg than placebo achieved MCS remission, MCS remission (alternative definition), and histologic remission. Treatment differences compared with placebo were numerically larger in the filgotinib 200 mg group than in the filgotinib 100 mg group.
The proportions of biologic-experienced Japanese patients who achieved key efficacy outcomes at week 10 were low across treatment groups (Fig. 1) with the exception of histologic remission, which was achieved by numerically high proportions of patients in the filgotinib groups (Fig. 1J). At week 58, numerically higher proportions of Japanese patients in the filgotinib 200 mg and 100 mg groups than the respective placebo groups achieved 6-month corticosteroid-free clinical remission, MCS remission, MCS remission (alternative definition), endoscopic remission, and histologic remission (Fig. 2). Among patients who received corticosteroids before enrollment, 6-month corticosteroid-free remission was achieved by 11.1% (1/9) of those in the filgotinib 200 mg group and by 0% (0/4) of those in the respective placebo group (Δ 11.1%, 95% CI, −27.5% to 49.7%), and by 33.3% (2/6) of those in the filgotinib 100 mg group and by 0% (0/1) of those in the respective placebo group (Δ 33.3%, 95% CI, −62.7% to 100.0%). Differences between treatment and placebo were numerically larger in the filgotinib 200 mg group than in the filgotinib 100 mg group in clinical remission, histologic remission, and MCS remission at week 58 in both biologic-naïve patients and biologic-experienced patients (Supplementary Fig. 4).
3) Exploratory Endpoints in SELECTION
Among both biologic-naïve and biologic-experienced Japanese patients, numerically greater proportions of those treated with filgotinib 200 mg or 100 mg achieved an MCS response at week 10 than those who received placebo (Supplementary Fig. 5A and B). At week 58, numerically higher proportions of Japanese patients achieved an MCS response in the filgotinib 200 mg group than in the respective placebo group (Supplementary Fig. 5C).
In both induction studies A and B, numerically greater reductions in pMCS from baseline over time from week 2 were reported for Japanese patients treated with filgotinib 200 mg or 100 mg than with placebo (Supplementary Fig. 6A and B). Reductions in pMCS were numerically greatest in patients treated with filgotinib 200 mg. In the maintenance study, Japanese patients in the filgotinib 200 mg or 100 mg groups experienced sustained improvements in pMCS versus maintenance baseline, while increases in pMCS were observed in the respective placebo groups (Supplementary Fig. 6C).
A numerically higher proportion of biologic-naïve Japanese patients who received filgotinib 200 mg achieved endoscopic response (a component of the primary endpoint) at week 10 than those who received placebo (33.3% [5/15] vs. 0% [0/6], Δ 33.3%, 95% CI, −2.2% to 68.9%) (Supplementary Fig. 7A). The proportions of biologic-experienced Japanese patients who achieved endoscopic response at week 10 were low across treatment groups (filgotinib 200 mg, 3.4% [1/29]; filgotinib 100 mg, 10.3% [3/29]; placebo, 7.1% [1/14]) (Supplementary Fig. 7B). In the maintenance study, a numerically higher proportion of Japanese patients in the filgotinib 200 mg group achieved endoscopic response at week 58 than in the respective placebo group (25.0% [5/20] vs. 11.1% [1/9], Δ 13.9%, 95% CI, −22.1% to 49.9%) (Supplementary Fig. 7C).
4) Symptomatic Remission
Numerically higher proportions of biologic-naïve and biologicexperienced Japanese patients who received filgotinib 200 mg achieved symptomatic remission (another component of the primary endpoint) at week 10 than those who received placebo (biologic-naïve, 60.0% [9/15] vs. 16.7% [1/6], Δ 43.3%, 95% CI, −7.1% to 93.8%; biologic-experienced, 48.3% [14/29] vs. 7.1% [1/14], Δ 41.1%, 95% CI, 13.2% to 69.1%) (Supplementary Fig. 8A and B). In the maintenance study, a numerically higher proportion of Japanese patients in the filgotinib 200 mg group achieved symptomatic remission at week 58 than in the respective placebo group (60.0% [12/20] vs. 0% [0/9], Δ 60.0%, 95% CI, 30.5% to 89.5%) (Supplementary Fig. 8C).
3. Safety
In induction studies A and B combined, mean ± standard deviation (SD) duration of exposure to the study drug was 10.8 ± 1.2 weeks, 10.9 ± 1.0 weeks, and 9.4 ± 3.6 weeks for patients treated with filgotinib 200 mg, filgotinib 100 mg, and placebo, respectively. In the maintenance study, mean ± SD duration of exposure to the study drug was 38.2 ± 16.9 weeks in the filgotinib 200 mg group and 25.3 ± 15.7 weeks in the respective placebo group; 34.0 ± 16.8 weeks in the filgotinib 100 mg group and 27.9 ± 20.6 weeks in the respective placebo group; and 27.0 ± 19.0 weeks in the induction placebo/maintenance placebo group.
The proportions of Japanese patients who experienced AEs were similar across treatment groups in both the induction studies (Table 2) and the maintenance study (Table 3). In the induction studies, the most commonly reported AEs were nasopharyngitis and UC; in the maintenance study, the most commonly reported AEs were nasopharyngitis, UC, and influenza. In the induction studies, AEs leading to study drug discontinuation occurred in 3 patients (15.0%) in the placebo group, and in 1 patient in both the filgotinib 100 mg (2.2%) and 200 mg (2.3%) groups. In the maintenance study, 2 patients (14.3%) in the filgotinib 100 mg group experienced AEs that led to study drug discontinuation.
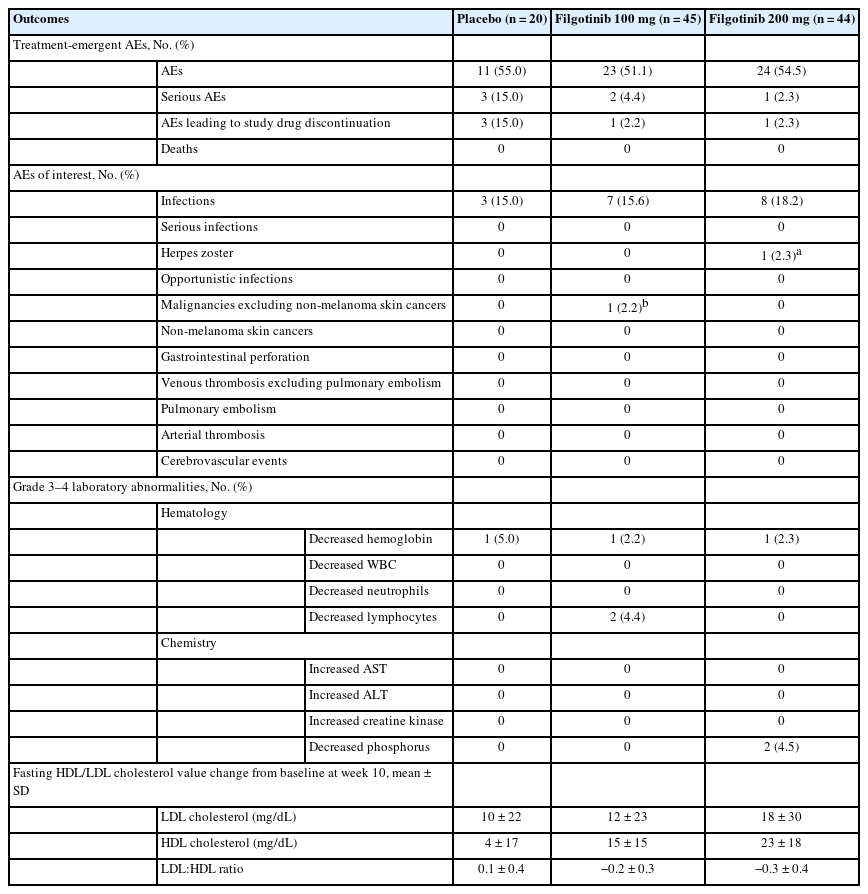
Summary of Safety Outcomes in Japanese Patients in Induction Studies A and B Combined
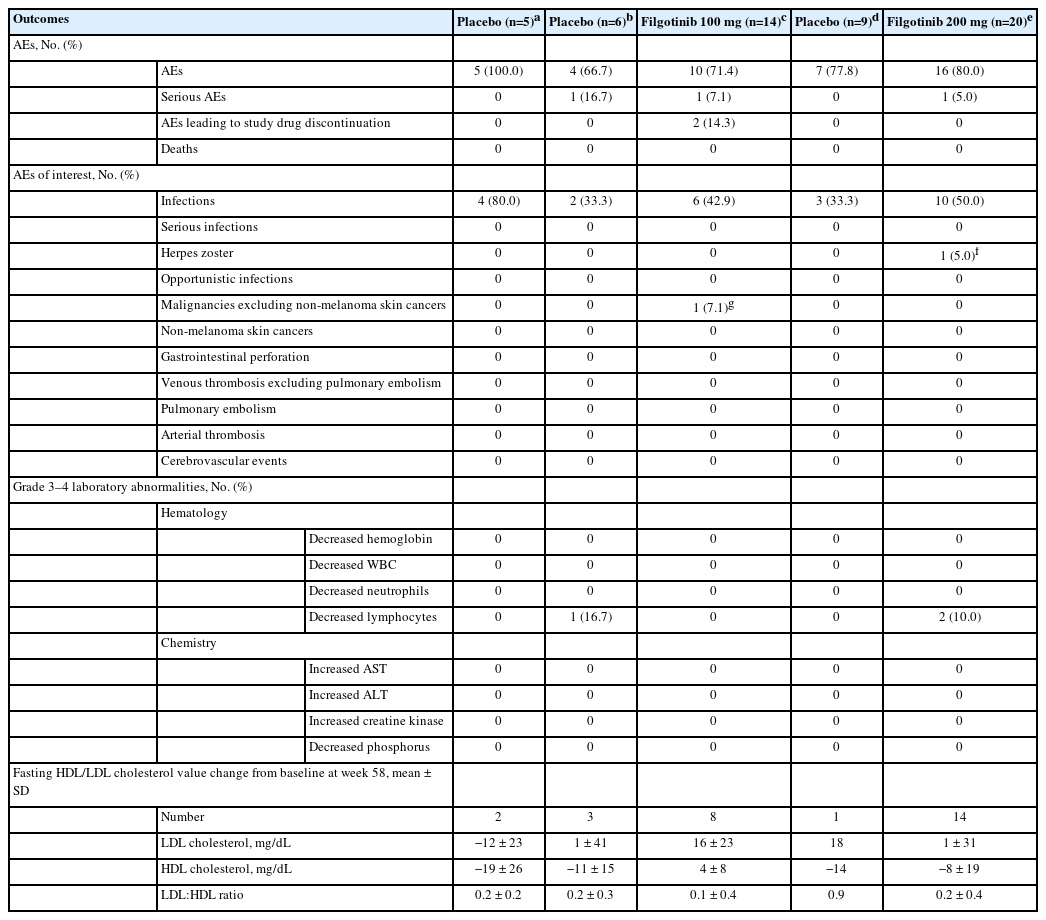
Summary of Safety Outcomes in Japanese Patients in the Maintenance Study
In the induction studies, serious AEs (SAEs) were reported in 6 Japanese patients (Table 2): 3 patients in the placebo group (UC, n = 2; acute pancreatitis, n = 1), 2 patients in the filgotinib 100 mg group (anemia, n = 1; vomiting, n = 1), and 1 patient in the filgotinib 200 mg group (intervertebral disc protrusion). In the maintenance study, 3 Japanese patients experienced SAEs (Table 3): 1 patient in the filgotinib 100 mg group (colon cancer), 1 patient in the induction filgotinib 100 mg/maintenance placebo group (meniscus injury), and 1 patient in the filgotinib 200 mg group (pyrexia). No Japanese patients died during the trial.
In the induction studies, infections occurred in 8 patients (18.2%) in the filgotinib 200 mg group, 7 patients (15.6%) in the filgotinib 100 mg group, and 3 patients (15.0%) in the placebo group (Table 2). In the maintenance study, infections were reported in 10 patients (50.0%) in the filgotinib 200 mg group and 3 patients (33.3%) in the respective placebo group, and in 6 patients (42.9%) in the filgotinib 100 mg group and 2 patients (33.3%) in the respective placebo group (Table 3). A herpes zoster infection was reported in 1 patient, a 56-year-old man, in the induction study A filgotinib 200 mg group, and in another patient, a 62-year-old woman, in the maintenance study filgotinib 200 mg group; both infections were nonserious and grade 2 in severity. No opportunistic or serious infections were reported for Japanese patients in the induction or maintenance studies.
No Japanese patients experienced gastrointestinal perforations, non-melanoma skin cancers, venous thromboses, or pulmonary embolisms in the induction or maintenance studies. One Japanese patient, a 64-year-old man, in the maintenance study filgotinib 100 mg group who had participated in induction study A had grade 3 colon cancer (carcinoma in the large intestine). The cancer was initially reported during induction when mucosal abnormalities were detected via a colonoscopy. Further investigation including endoscopic submucosal dissection led to the diagnosis of colon cancer during the maintenance study. The diagnosis was deemed by the investigator not to be related to the study treatment.
The proportions of patients with grade 3 or 4 laboratory abnormalities were low and generally similar across treatment groups in all 3 studies.
DISCUSSION
In these post-hoc analyses, we evaluated the safety and efficacy of filgotinib among Japanese patients with moderately to severely active UC enrolled in Japan in the phase 2b/3 SELECTION trial. The data presented here suggest that filgotinib 200 mg may be efficacious in inducing clinical remission in biologic-naïve Japanese patients. In biologic-experienced Japanese patients, a treatment effect was observed for MCS response, pMCS reduction, and histologic remission, but not clinical remission owing to the low rates of remission reported across treatment groups. In the maintenance study, clinical remission was achieved by numerically higher proportions of biologic-naïve and biologic-experienced patients treated with filgotinib 200 mg than with placebo. In addition, both the filgotinib 200 mg and 100 mg doses were well tolerated, and no new safety findings were noted in the Japanese population.
During induction, numerically higher proportions of biologic-naïve patients treated with filgotinib 200 mg achieved clinical remission at week 10 than patients treated with placebo. The proportion of biologic-experienced patients in clinical remission at week 10 was low across treatment groups. It is noteworthy, though, that over half (62.1%) of the biologic-experienced patients treated with filgotinib 200 mg achieved an MCS response, compared with 14.3% of patients treated with placebo. Symptomatic remission was achieved by numerically greater proportions of biologic-experienced patients treated with either dose of filgotinib than placebo at week 10. In addition, improvements in UC symptoms (measured using pMCS) from week 2 (the first post-baseline assessment) were seen in both biologic-naïve and experienced patients upon treatment with filgotinib 200 mg. Improved symptoms were sustained in patients who continued to receive filgotinib 200 mg compared with those who received placebo in the maintenance study. These results are encouraging, because symptomatic improvement (including alleviation of rectal bleeding and high stool frequency symptoms) is a short-term goal of the recommended treat-to-target approach for UC, as described in the recent Selecting Therapeutic Targets in Inflammatory Bowel Disease II guidelines [24]. Conversely, endoscopic response was achieved by a low proportion of biologic-experienced patients across treatment groups, consistent with the results for clinical remission. Given that an MES of 0 or 1 was required for both clinical remission and endoscopic response, together these findings imply that endoscopic subscore was the most difficult subscore to modulate. Proportionally more Japanese patients had an endoscopic subscore of 3 at baseline than in the overall SELECTION population. This could imply more severe disease in Japanese patients and, given our stringent definition of clinical remission, this suggests proportionally more of these patients needed at least a 2-point reduction in endoscopic subscore to achieve week 10 clinical remission. Hence, this could have contributed to the low induction clinical remission rate in this subgroup. Nevertheless, given the small patient numbers, data should be interpreted with caution.
It is noteworthy that histologic remission was achieved by numerically greater proportions of both biologic-naïve and biologic-experienced filgotinib 200 mg-treated patients than placebo-treated patients at week 10. This is a promising result, given the stringent definition of histologic remission used in this trial. Histologic remission rates were also numerically higher in biologic-experienced patients than clinical or endoscopic remission rates. Clinical, endoscopic, and histological indices used to assess UC disease activity provide distinct information, and the absolute response rate for each associated endpoint varies depending upon its definition and stringency. Indeed, different histologic and endoscopic scoring systems have been shown to infer varying disease activity [25]. Furthermore, higher rates of histologic improvement versus clinical and endoscopic outcomes were reported in the phase 3 UC trial of ustekinumab [12]. Therefore, the rates of histologic remission observed in this trial are not unexpected, and may suggest a beneficial effect of filgotinib on this outcome.
In the maintenance study, clinical remission was achieved by a numerically higher proportion of patients treated with filgotinib 200 mg than with placebo. A treatment effect was observed for both biologic-naïve and biologic-experienced patients. Similar results were observed for histologic remission, MCS remission, and MCS remission per the alternative definition. It is noteworthy that clinical remission was achieved by nearly a quarter (23.1%) of biologic-experienced patients who responded to the induction treatment and received filgotinib 200 mg therapy during maintenance. In both the induction and maintenance studies, filgotinib 100 mg was generally less efficacious than filgotinib 200 mg, as was the case for the overall SELECTION population [23], supporting the use of the 200 mg dose.
Both doses of filgotinib were well tolerated among Japanese patients in the SELECTION trial, and AEs, SAEs, and AEs leading to discontinuation of the study drug were reported for similar proportions of patients across treatment groups in all 3 studies. Herpes zoster infections were experienced by 1 Japanese patient who received filgotinib 200 mg during induction and another who received filgotinib 200 mg during maintenance. Asian patients have been shown to have a higher risk of experiencing herpes zoster during treatment with JAK inhibitors than non-Asian patients [26,27]. In this post-hoc analysis, no dose-related differences in the incidence of herpes zoster were indicated in the induction or maintenance studies, nor was an increased incidence of herpes zoster observed over time in the maintenance study. In addition, no opportunistic infections, pulmonary embolisms, or venous thromboses were reported. One patient treated with filgotinib 100 mg during induction and maintenance had grade 3 colon cancer; the diagnosis was judged by the investigator not to be related to the study treatment. Overall, no new safety signals or trends were identified in Japanese patients compared with the total SELECTION population. In September 2021, the U.S. Food and Drug Administration announced that warnings about increased risk of serious heart-related events, cancer, blood clots, and death need to be provided for JAK inhibitors that treat certain chronic inflammatory conditions based on their review of a large randomized safety clinical trial of tofacitinib in patients with rheumatoid arthritis [28]. In SELECTION, the incidence of such events was similar among patients treated with filgotinib and placebo in both the Japanese and overall populations [23]. The overall good safety profile of filgotinib reported here may be a result of preferential inhibition of JAK1 over JAK2, JAK3, and TYK2 [17-19], though the presented data should be interpreted with caution owing to the relatively small number of Japanese patients enrolled in this trial.
Following observations of impaired spermatogenesis and histopathological changes to the testes and epididymis in rats and dogs during preclinical toxicology studies [29], 2 studies were designed to evaluate the potential effect of filgotinib on adult male semen parameters: 1 in men with moderately to severely active UC or Crohn’s disease (MANTA, ClinicalTrials.gov ID: NCT03201445), and the other in men with active rheumatic diseases (MANTA-RAy, ClinicalTrials.gov ID: NCT03926195). These studies are still ongoing, but in an interim analysis of pooled data from both studies for the primary endpoint, at week 13, 6.7% (8/120) and 8.3% (10/120) of participants treated with filgotinib 200 mg and placebo, respectively, experienced a ≥ 50% decrease from baseline in sperm concentration [30,31].
Strengths of the SELECTION trial included the simple dosing regimen, with no requirement for dose modification. Filgotinib has a low potential for drug–drug interactions [32], and the oral administration route negates the need for clinic visits for infusion or injections. In addition, no therapeutic drug monitoring is necessary to maintain optimal drug levels [32]. These attributes, in addition to the presented safety and efficacy data, suggest that filgotinib might be a viable treatment option for patients with moderately to severely active UC in Japan.
A potential limitation of the SELECTION trial is the relatively short duration of the study, and a separate long-term extension study (SELECTIONLTE; ClinicalTrials.gov ID: NCT02914535) is currently underway. The main limitation of these post-hoc analyses is the small number of Japanese patients. Therefore, we present descriptive statistics rather than statistical significance, and hence the results should be interpreted with caution. Collection and evaluation of long-term real-world data may give further insights into the effectiveness and safety of filgotinib for the treatment of UC in Japanese patients.
In conclusion, filgotinib was well tolerated and had a good safety profile among Japanese patients in the phase 2b/3 SELECTION trial. The results presented here, together with those of the overall population, suggest the potential of filgotinib 200 mg as a viable treatment option for patients with moderately to severely active UC in Japan. Owing to the small number of Japanese patients included in these analyses, the presented data should be interpreted with caution.
Notes
Funding Source
The SELECTION trial was sponsored by Gilead Sciences, Inc. (Foster City, CA, USA), and co-funded by Gilead Sciences, Inc. and Galapagos (Mechelen, Belgium).
Conflict of Interest
Hibi T has received lecture fees from Aspen Japan K.K., Janssen, JIMRO, Mitsubishi Tanabe Pharma, Mochida Pharmaceutical, Pfizer, and Takeda Pharmaceutical; research grants from AbbVie GK, Activaid, Alfresa Pharma Corporation, Bristol Myers Squibb, Eli Lilly Japan K.K., Ferring Pharmaceuticals, Gilead Sciences, Inc., Janssen Pharmaceutical K.K., JMDC Inc., Mochida Pharmaceutical Co., Ltd., Nippon Kayaku Co., Ltd., Pfizer Japan Inc., and Takeda Pharmaceutical Co., Ltd.; scholarship contributions from Mitsubishi Tanabe Pharma Corporation, Nippon Kayaku Co., Ltd., and Zeria Pharmaceutical Co., Ltd.; and study group sponsorship from AbbVie GK, EA Pharma Co., Ltd., JIMRO Co., Ltd., Kyorin Pharmaceutical Co., Ltd., Mochida Pharmaceutical Co., Ltd., Otsuka Holdings, and Zeria Pharmaceutical Co., Ltd.
Motoya S has received lecture fees from Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corporation, and Takeda Pharmaceutical Co., Ltd.; and research grants from AstraZeneca K.K., EA Pharma Co., Ltd., Eli Lilly Japan K.K., Janssen Pharmaceutical K.K., Pfizer Inc., and Takeda Pharmaceutical Co., Ltd.
Hisamatsu T has a joint research agreement with Alfresa Pharma Co., Ltd. and EA Pharma Co., Ltd.; and has received grant support from AbbVie GK, Daiichi Sankyo, EA Pharma Co., JIMRO Co., Ltd., Kyorin Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Ltd., Mochida Pharmaceutical Co., Ltd., Nippon Kayaku Co., Ltd., Pfizer Inc., Takeda Pharmaceutical Co., Ltd., and Zeria Pharmaceutical Co., Ltd.; and consulting and lecture fees from AbbVie GK, Celgene K.K., Ltd., EA Pharma Co., Janssen Pharmaceutical K.K., JIMRO Co., Kyorin Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., Pfizer Inc., and Takeda Pharmaceutical Co., Ltd.
Hirai F has received lecture fees from AbbVie GK, EA Pharma Co., Ltd., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., and Takeda Pharmaceutical Co., Ltd.; research grants from Eli Lilly Japan K.K. and Janssen Pharmaceutical K.K.; scholarship contributions from AbbVie GK, Ayumi Pharmaceutical Corporation, EA Pharma Co., Ltd., Eisai Co., Ltd., Kissei Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., and Otsuka Pharmaceutical Co., Ltd.; and study group sponsorship from AbbVie GK, EA Pharma Co., Ltd., JIMRO Co., Ltd., Kyorin Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, and Zeria Pharmaceutical Co., Ltd.
Watanabe K has received lecture fees from AbbVie GK, EA Pharma Co., Ltd., Kissei Pharmaceutical Co., Ltd., Kyorin Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., Pfizer Inc., and Takeda Pharmaceutical Co., Ltd.; research grants from EA Pharma Co., Ltd., EP-CRSU Co., Ltd., and Takeda Pharmaceutical Co., Ltd.; scholarship contributions from AbbVie GK, EA Pharma Co., Ltd., JIMRO Co., Mitsubishi Tanabe Pharma Corporation, and Nippon Kayaku Co., Ltd.; and study group sponsorship from AbbVie GK, Asahi Kasei Medical Co., Ltd., EA Pharma Co., Ltd., JIMRO Co., Kyorin Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Factory Inc., and Zeria Pharmaceutical Co., Ltd.
Matsuoka K has served as a scientific adviser for EA Pharma; has served on advisory boards for Boehringer Ingelheim, Bristol Myers Squibb, and Eli Lilly and Company; and has received personal fees from AbbVie, EA Pharma, Janssen, JIMRO, Kissei Pharmaceutical, Kyorin Pharmaceutical, Mitsubishi Tanabe Pharma, Mochida Pharmaceutical, Pfizer Inc, Takeda Pharmaceutical, and Zeria Pharmaceutical; and research grants from AbbVie, EA Pharma, JIMRO, Kissei Pharmaceutical, Kyorin Pharmaceutical, Mitsubishi Tanabe Pharma, Mochida Pharmaceutical, Nippon Kayaku, and Takeda Pharmaceutical.
Saruta M has received consulting and lecture fees from EA Pharma Co., Ltd., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Co., Ltd., and Takeda Pharmaceutical Co., Ltd.; and research grants from EA Pharma Co., Ltd., EP-CRSU Co., Ltd., Mitsubishi Tanabe Pharma, Mochida Pharmaceutical Co., Ltd., and Zeria Pharmaceutical Co., Ltd.
Kobayashi T has received research grants from AbbVie GK, Activaid, Alfresa Pharma Corporation, Bristol Myers Squibb, Eli Lilly Japan K.K., Ferring Pharmaceuticals, Gilead Sciences, Inc., Janssen Pharmaceutical K.K., JMDC Inc., Mochida Pharmaceutical Co., Ltd., Nippon Kayaku Co., Ltd., Pfizer Japan Inc., and Takeda Pharmaceutical Co., Ltd.; scholarship contributions from Mitsubishi Tanabe Pharma Corporation, Nippon Kayaku Co., Ltd., and Zeria Pharmaceutical Co., Ltd.; and study group sponsorship from AbbVie GK, EA Pharma Co., Ltd., JIMRO Co., Ltd., Kyorin Pharmaceutical Co., Ltd., Mochida Pharmaceutical Co., Ltd., Otsuka Holdings, and Zeria Pharmaceutical Co., Ltd.
Feagan BG has received consulting and/or speaker fees from AbbVie, AdMIRx, AgomAB Therapeutics, Akebia, Alivio Therapeutics, Allakos, Amgen, Applied Molecular Transport Inc., Arena Pharma, Avir, Azora Therapeutics, Boehringer Ingelheim, Boston Pharma, Celgene/BMS, Connect BioPharma, Cytoki, Disc Medicine, Eli Lilly, Equillium, Everest Clinical Research Corp., Ferring, Galapagos, Galen Atlantica, Genentech/Roche, Gilead, Glenmark, Gossamer Pharma, GSK, Hoffmann-La Roche, Hot Spot Therapeutics, ImmuNext, Index Pharma, Intact Therapeutics, Janssen, Japan Tobacco Inc., Kaleido Biosciences, Leadiant, Millennium, MiroBio, Morphic Therapeutics, Mylan, OM Pharma, Origo BioPharma, Otsuka, Pandion Therapeutics, Pfizer, Progenity, Prometheus Therapeutics and Diagnostics, PTM Therapeutics, Q32 Bio, Rebiotix, RedHill Biopharma, REDX, Sandoz, Sanofi, Seres Therapeutics, Surrozen Inc., Takeda, Teva, Thelium, Theravance, Tigenix, Tillotts, UCB Pharma, VHsquared Ltd., Viatris, Ysios, and Zealand Pharma.
Tasset C and Besuyen R are employees and shareholders of Galapagos.
Yun C, Crans G, and Zhang J are employees and shareholders of Gilead Sciences, Inc.
Kondo A is an employee of Gilead Sciences K.K. and shareholder of Gilead Sciences, Inc.
Watanabe M has received grants from Alfresa Pharma Corporation, JIMRO Co., Ltd., Kyorin Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., and Miyarisan Pharmaceutical Co., Ltd.; grants and personal fees from AbbVie GK, Astellas Pharma Inc., EA Pharma Co., Ltd., Kissei Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., Nippon Kayaku Co., Ltd., Pfizer Japan Inc., Takeda Pharmaceutical Co., Ltd., and Zeria Pharmaceutical Co., Ltd.; and personal fees from Celgene K.K., Celltrion Healthcare Co., Ltd., Eli Lilly Japan K.K., Gilead Sciences, Inc., and Janssen Pharmaceutical K.K.
Hibi T, Matsuoka K, and Watanabe M are editorial board members of the journal but were not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Data Availability Statement
Anonymized individual patient data will be shared upon request for research purposes dependent upon the nature of the request, the merit of the proposed research, the availability of the data, and its intended use. The full data-sharing policy for Gilead Sciences, Inc., can be found at https://www.gilead.com/about/ethics-and-code-of-conduct/policies.
Author Contribution
Conceptualization: Tasset C, Yun C. Data curation: all authors. Formal analysis: Zhang J, Crans G. Investigation: Hibi T, Motoya S, Hisamatsu T, Hirai F, Watanabe K, Matsuoka K, Saruta M, Kobayashi T, Feagan BG, Watanabe M. Writing - review & editing: all authors. Approval of final manuscript: all authors.
Others
Medical writing support for the preparation of this manuscript was provided by Frances Thompson, PhD, of PharmaGenesis London, London, UK, and funded by Gilead Sciences K.K. (Tokyo, Japan).
Supplementary Material
Supplementary materials are available at the Intestinal Research website (https://www.irjournal.org).
Supplementary Fig. 1. Study design. Clinical remission was defined as an MES of 0 or 1, rectal bleeding subscore of 0, and at least a 1-point decrease in stool frequency from induction baseline to achieve a subscore of 0 or 1. MCS response was defined as a reduction of at least 3 points in MCS and at least 30% from induction baseline with an accompanying decrease in rectal bleeding subscore of at least 1 point, or an absolute rectal bleeding subscore of 0 or 1. aResponders were patients who achieved clinical remission or MCS response at week 10. MCS, Mayo Clinic score; MES, Mayo endoscopic subscore.
ir-2021-00143-Supplementary-Fig-1.pdfSupplementary Fig. 2. Disposition of Japanese patients in induction study A (biologic-naïve patients) (A) and induction study B (biologicexperienced patients) (B).
ir-2021-00143-Supplementary-Fig-2.pdfSupplementary Fig. 3. Disposition of Japanese patients in the maintenance study.
ir-2021-00143-Supplementary-Fig-3.pdfSupplementary Fig. 4. Proportion of biologic-naïve and biologic-experienced Japanese patients achieving clinical remission (A, B), 6-month corticosteroid-free clinical remission (C, D), sustained clinical remission (E, F), MCS remission (G, H), MCS remission (alternative definition) (I, J), endoscopic remission (K, L), and histologic remission (M, N) in the maintenance study at week 58. Clinical remission was defined as an MES of 0 or 1, rectal bleeding subscore of 0, and at least a 1-point decrease in stool frequency from induction baseline to achieve a subscore of 0 or 1. Six-month corticosteroid-free clinical remission was defined as clinical remission with no corticosteroid use for the indication of UC for at least 6 months before week 58 among patients who were receiving corticosteroids at the baseline of the maintenance study. Sustained clinical remission was defined as clinical remission at both week 10 and week 58. MCS remission was defined as a total MCS of not more than 2 and no single subscore greater than 1. MCS remission (alternative definition) was defined as rectal bleeding, stool frequency, and physician’s global assessment subscores of 0; an endoscopic subscore of 0 or 1; and an overall MCS of 1 or 0. Endoscopic remission was defined as an MES of 0. Histologic remission was defined as grade 0 Geboes score of ≤0.3, grade 1 score of ≤1.1, grade 2A score of ≤2A.3, grade 2B score of 2B.0, grade 3 score of 3.0, grade 4 score of 4.0, and grade 5 score of 5.0. MCS, Mayo Clinic score; MES, Mayo endoscopic subscore; UC, ulcerative colitis.
ir-2021-00143-Supplementary-Fig-4.pdfSupplementary Fig. 5. Proportion of Japanese patients who achieved an MCS response in induction studies A and B (A, B) at week 10, and in the maintenance study at week 58 (C). Error bars indicate 95% confidence intervals (CIs). CIs for 0-responder treatment arms are not presented. 95% CIs were calculated based on normal approximation with a continuity correction. MCS response was defined as a reduction of at least 3 points in MCS and at least 30% from induction baseline with an accompanying decrease in rectal bleeding subscore of at least 1 point, or an absolute rectal bleeding subscore of 0 or 1. MCS, Mayo Clinic score.
ir-2021-00143-Supplementary-Fig-5.pdfSupplementary Fig. 6. Change in pMCS over time in Japanese patients in induction study A (A), induction study B (B), and the maintenance study (C). pMCS was defined as the sum of Mayo rectal bleeding, stool frequency, and physician’s global assessment subscores. pMCS, partial Mayo Clinic score.
ir-2021-00143-Supplementary-Fig-6.pdfSupplementary Fig. 7. Proportion of Japanese patients who achieved endoscopic response in induction studies A and B at week 10 (A, B), and in the maintenance study at week 58 (C). Error bars indicate 95% confidence intervals (CIs). CIs for 0-responder treatment arms are not presented. 95% CIs were calculated based on normal approximation with a continuity correction. Endoscopic response was defined as an MES of 0 or 1. MES, Mayo endoscopic subscore.
ir-2021-00143-Supplementary-Fig-7.pdfSupplementary Fig. 8. Proportion of Japanese patients who achieved symptomatic remission in induction studies A and B at week 10 (A, B), and in the maintenance study at week 58 (C). Error bars indicate 95% confidence intervals (CIs). CIs for 0-responder treatment arms are not presented. 95% CIs were calculated based on normal approximation with a continuity correction. Symptomatic remission was defined as having a Mayo rectal bleeding subscore of 0, and at least a 1-point decrease in stool frequency subscore from induction baseline to achieve a subscore of 0 or 1.
ir-2021-00143-Supplementary-Fig-8.pdfSupplementary Table 1. Baseline Demographics and Characteristics of Japanese Patients in the Maintenance Study
ir-2021-00143-Supplementary-Table-1.pdf