Efficacy and safety of mirikizumab as induction and maintenance therapy for Japanese patients with moderately to severely active ulcerative colitis: a subgroup analysis of the global phase 3 LUCENT-1 and LUCENT-2 studies
Article information

Abstract
Background/Aims
Mirikizumab is a p19-directed anti-interleukin-23 antibody with potential efficacy against ulcerative colitis (UC). We evaluated the efficacy and safety of mirikizumab in a Japanese subpopulation with moderately to severely active UC from the LUCENT-1 and LUCENT-2 studies.
Methods
LUCENT-1 and LUCENT-2 were phase 3, randomized, double-blind, placebo-controlled trials of mirikizumab therapy in adults with moderately to severely active UC. LUCENT-1 was a 12-week induction trial where patients were randomized 3:1 to receive intravenous mirikizumab 300 mg or placebo every 4 weeks (Q4W). Patients achieving a clinical response with mirikizumab following the induction study were re-randomized 2:1 to double-blind treatment with either mirikizumab 200 mg or placebo subcutaneously Q4W during the 40-week maintenance study. The primary outcomes were clinical remission at week 12 of LUCENT-1 and week 40 of LUCENT-2.
Results
A total of 137 patients enrolled in Japan were randomized to mirikizumab (n = 102) or placebo (n = 35). Compared with placebo, patients who received mirikizumab showed numerically higher clinical remission at week 12 of induction (32.4% [n = 33] vs. 2.9% [n = 1]) and at week 40 of maintenance (48.9% [n = 23] vs. 28.0% [n = 7]). A greater number of patients achieved key secondary endpoints in the mirikizumab group compared with placebo. The frequency of treatment-emergent adverse events was similar across mirikizumab and placebo groups. Efficacy and safety results observed in the Japanese subpopulation were generally consistent with those in the overall population.
Conclusions
Mirikizumab induction and maintenance treatments were effective in Japanese patients with moderately to severely active UC. No new safety concerns were identified.
INTRODUCTION
Ulcerative colitis (UC) is a chronic relapsing disease characterized by mucosal inflammation of the colon [1,2]. Typical symptoms include rectal bleeding, diarrhea, and bowel urgency which negatively impact patients’ quality of life [3]. UC is classed as a global disease, and although the incidence is stabilizing in Western countries [4], the prevlence of UC has risen substantially in Japan over the previous 2 decades [5].
The goal of treatment for UC is to induce and maintain remission. Despite significant treatment advances, current therapies pose limitations and/or lose clinical efficacy over time [6]. Hence, a need remains for new therapeutics with an adequate benefit: risk profile to manage this complex disease.
The interleukin (IL)-23 axis plays a significant role in the pathogenesis of UC [7] and has thus become an important target for drug development. IL-23 contains a unique p19 subunit and a common p40 subunit which it shares with IL-12 [8]. Mirikizumab is a humanized immunoglobulin G4–variant monoclonal antibody that specifically binds to the p19 subunit of IL-23. In phase 2 trials, mirikizumab demonstrated efficacy in patients with moderately to severely active UC, including in Japanese patients [9,10].
We recently reported the findings from phase 3, randomized, global, double-blind, placebo-controlled studies for induction (LUCENT-1, NCT03518086) and maintenance (LUCENT-2, NCT03524092) treatment with mirikizumab in UC [11]. These were the first reported phase 3 studies describing the safety and efficacy of targeting the p19 subunit of IL-23 for the treatment of UC. LUCENT-1 and LUCENT-2 demonstrated mirikizumab was more effective compared with placebo in inducing and maintaining clinical remission in patients with moderately to severely active UC, and were the first phase 3 studies in UC to evaluate bowel urgency and demonstrate the efficacy of mirikizumab in improving bowel urgency severity and bowel urgency numeric rating scale (NRS) of 0 or 1 [11]. The frequency of treatment-emergent adverse events (TEAEs) was similar across mirikizumab and placebo groups, with no new major safety concerns identified [11]. In the current study, we describe efficacy and safety of mirikizumab compared with placebo in Japanese patients with moderately to severely active UC from the phase 3 LUCENT-1 and LUCENT-2 trials.
METHODS
1. Study Design
LUCENT-1 and LUCENT-2 were both phase 3, multicenter, double-blind, randomized, parallel-arm, placebo-controlled trials. LUCENT-1 was a 12-week induction trial and LUCENT-2 was a 40-week maintenance trial, comprising a total of 52 weeks of treatment. The study design has been previously described [9] and is shown in Supplementary Fig. 1. Study design modifications specific to Japanese patients are outlined in the Supplementary Material.
Briefly, in LUCENT-1, eligible patients were randomized 3:1 to receive mirikizumab 300 mg or placebo via intravenous (IV) infusion every 4 weeks (Q4W) at weeks 0, 4, and 8.
Randomization was stratified by biologic or tofacitinib-failure status (yes/no), baseline corticosteroid use (yes/no), baseline disease activity (modified Mayo score [4–6] or [7–9]), and region (North America/Europe/other). Assignment to treatment groups was determined by a computer-generated random sequence using an interactive web-response system. Anti-tumor necrosis factor antibodies and Janus kinase inhibitors were required to be discontinued at least 8 weeks and at least 4 weeks prior to the screening endoscopy, respectively, and were subsequently prohibited throughout the duration of the study. As per the study design, only patients who achieved a clinical response to mirikizumab treatment at the end of the induction study (LUCENT-1) were randomized 2:1 to doubleblind treatment with either mirikizumab 200 mg subcutaneously (SC) Q4W or placebo SC Q4W during the maintenance study (LUCENT-2). Patients who did not achieve a clinical response at the end of the induction study were not included in the double-blind treatment arms during the maintenance study.
Randomization in LUCENT-2 was stratified by biologic or tofacitinib-failure status (yes/no), induction remission status (yes/no), corticosteroid use (yes/no), and region (North America/Europe/other). Patients who achieved a clinical response to placebo at the end of the induction study continued blinded placebo during the maintenance study. Induction clinical responders who experienced a loss of response to either mirikizumab or placebo at or after week 12 of the maintenance study could receive rescue induction therapy with open-label mirikizumab 300 mg IV Q4W for 3 doses. Patients with loss of response who, in the opinion of the investigator, had a clinical benefit from rescue therapy, were considered for transition to the long-term extension study. Patients without a clinical response to mirikizumab or placebo in the induction study were enrolled in the maintenance study as nonresponders, receiving open-label extended induction dosing with mirikizumab 300 mg IV Q4W for 3 doses. Those who achieved clinical response following extended induction dosing at week 12 of the maintenance study received open-label mirikizumab 200 mg SC Q4W maintenance dosing through week 40 while nonresponders discontinued the study.
The study was conducted in accordance with the International Conference on Harmonisation for Good Clinical Practice and the Declaration of Helsinki. The study protocols were approved by the institutional review board responsible for oversight at each center. All patients provided written informed consent to participate in the study.
2. Participants
Adults (aged 18–80 years) with moderately to severely active UC at screening (modified Mayo score of 4–9 [range 0 to 9] with an endoscopic subscore of 2–3 [range 0 to 3]) were eligible for inclusion in the LUCENT-1 induction study. Eligible patients had an established diagnosis of UC at least 3 months prior to baseline, with endoscopic evidence and histopathology to support the diagnosis, and UC extending beyond the rectum. Patients had inadequate response, loss of response, or intolerance to corticosteroids or immunomodulators (see Supplementary Material) or had failed or demonstrated an intolerance to a biologic medication or Janus kinase inhibitors. Patients were allowed to use oral 5-aminosalicyclic acid, immunomodulators azathioprine or 6-mercaptopurine, or methotrexate with stable doses required. Patients taking corticosteroids at baseline were required to maintain a stable dose during the induction trial but were required to begin corticosteroid taper upon entering the LUCENT-2 maintenance study as induction responders.
Patients were ineligible for study inclusion if UC was limited to the rectum, if they were diagnosed with other forms of inflammatory bowel disease or an immunodeficiency syndrome which may cause UC-like colonic inflammation, had extensive colonic resection, stricture/stenosis within the small bowel or colon, toxic megacolon, colonic adenoma that had not been removed, dysplasia of colonic mucosa, gastrointestinal cancer, or failed 3 or more biologic therapies (excluding tofacitinib) for UC. Further details of patient inclusion and exclusion criteria for LUCENT-1 and LUCENT-2 have been previously described, and criteria specific to Japanese patients are outlined in the Supplementary Appendix. Patients who completed the 12-week treatment period (LUCENT-1), irrespective of clinical response status, were eligible to participate in the 40-week maintenance study (LUCENT-2).
3. Efficacy and Safety Evaluation
For LUCENT-1, the primary endpoint was the proportion of patients in clinical remission at week 12. Clinical remission was based on the modified Mayo score and defined as: stool frequency subscore =0 (range 0 to 3), or stool frequency =1 with a ≥ 1-point decrease from baseline, rectal bleeding subscore =0 (range 0 to 3), and endoscopic subscore =0 or 1 (excluding friability). Key secondary endpoints at week 12 included alternative clinical remission, clinical response, endoscopic improvement, symptomatic remission (week 4 and week 12), clinical response in the biologic-failed population, histologic-endoscopic mucosal improvement (as demonstrated in colonic biopsy samples), and change from baseline in bowel urgency NRS.
For LUCENT-2, the primary endpoint was the proportion of patients in clinical remission at week 40 from patients who achieved a clinical response with mirikizumab in LUCENT-1. Key secondary endpoints at week 40 included alternative clinical remission among patients induced into clinical response in LUCENT-1, endoscopic improvement, maintenance of clinical remission, corticosteroid-free clinical remission, histologic-endoscopic mucosal improvement plus absence of neutrophils (as demonstrated in colonic biopsy samples), improvement in bowel urgency severity, and bowel urgency NRS of 0 or 1. Bowel urgency NRS of 0 or 1 indicates normal or near normalization of bowel urgency status.
An additional endpoint for LUCENT-1 and LUCENT-2 was the average serum concentration of mirikizumab (full details are outlined in Supplementary Table 1). A complete list of primary and secondary endpoints and their definitions for both LUCENT-1 and LUCENT-2 are provided in Supplementary Table 2.
Safety outcomes included TEAEs, study discontinuation due to adverse events (AEs), deaths, serious AEs, AEs of special interest, laboratory values, and vital signs. AEs of special interest included infections (opportunistic and serious), hepatic safety, hypersensitivity, infusion reactions, injection site reactions, cerebrocardiovascular events, malignancies, suicidal ideation, and depression.
4. Statistical Analyses
Efficacy and safety analyses were performed on the modified intent-to-treat populations of LUCENT-1 and LUCENT-2. These populations included all randomized patients who received at least 1 dose of study drug during the respective study. The results of the Japanese subpopulation analyses were summarized descriptively. Consistency in efficacy and safety outcomes between the Japanese subpopulation and overall population were evaluated.
For assessments of the primary endpoints and other cate gorical efficacy endpoints, the Cochran-Mantel-Haenszel test was used to compare the treatment groups while adjusting for stratification factors. For analysis of categorical efficacy and health outcomes variables, patients were considered nonresponders if they did not meet the categorical efficacy criteria, had missing clinical efficacy data at a time point of interest, or took rescue medication prior to the time point of interest. Treatment comparisons of continuous efficacy variables were made using the mixed-effects model for repeated measures analyses, with censoring of data following rescue therapy. Primary and major secondary endpoints for the overall population were tested after adjusting for multiplicity using a predefined graphical procedure with an overall family-wise error rate of 0.00125 for LUCENT-1 and 0.05 for LUCENT-2. For both studies, analyses of hypotheses without multiplicity control, including all analyses in the Japanese subpopulation, are reported with point estimates and 95% confidence intervals, without P-values; the widths of the confidence intervals are not adjusted for multiple testing and should not be used to infer definitive treatment effects.
RESULTS
1. Patients
In the induction study, of 1,162 patients, 137 participated from institutions located in Japan and were randomized to either mirikizumab 300 mg (n=102) or placebo (n=35). All 137 patients were Japanese, with 1 Japanese patient enrolled outside of Japan not included in this subpopulation analysis. Three patients in the mirikizumab arm discontinued study treatment due to AEs (colitis ulcerative, n=2) and lack of efficacy (n=1), while 9 patients discontinued from the placebo arm due to AEs (colitis ulcerative n=4, sinusitis n=1, arthritis n=1) and lack of efficacy (n=3). Overall, 99 Japanese patients in the mirikizumab arm and 26 patients in the placebo arm completed treatment through week 12. A total of 123 patients from LUCENT-1 entered the LUCENT-2 maintenance study. Of the 72 Japanese patients who achieved a clinical response to mirikizumab in the induction study, 47 were randomized to receive mirikizumab 200 mg and 25 were randomized to receive placebo (Fig. 1).
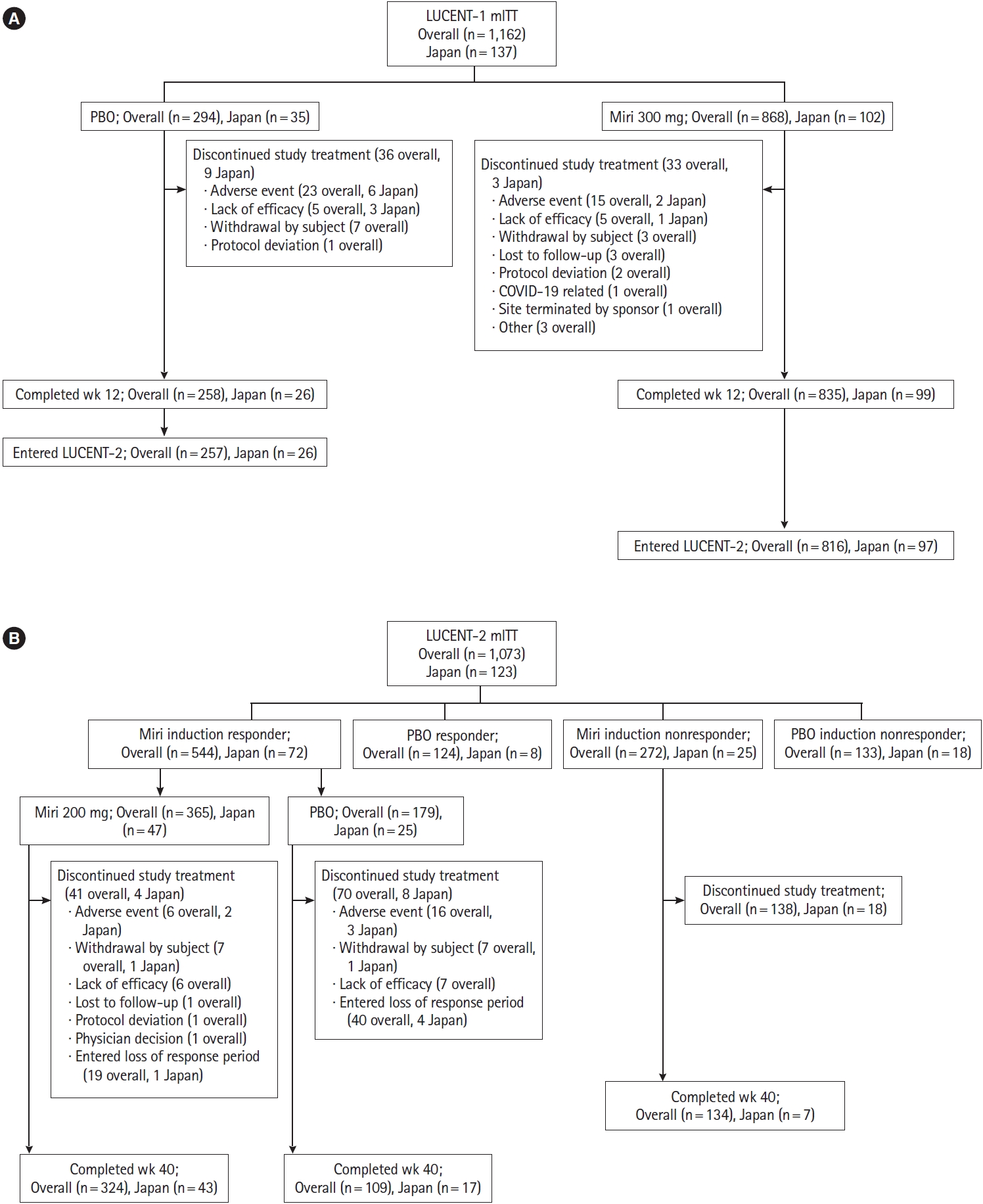
Study design and patient disposition in (A) LUCENT-1 induction and (B) LUCENT-2 maintenance trials. Miri, mirikizumab; mITT, modified intention-to-treat; PBO, placebo.
There were no meaningful imbalances in baseline demographic and disease characteristics between treatment arms in the overall and in the Japanese subpopulation. Body mass index and mean C-reactive protein were lower in Japanese patients than overall population and mean disease duration was longer in the Japanese subgroup. Japanese patients had lower baseline corticosteroid use but higher baseline use of immunomodulators than the overall population (Table 1). In addition, a slightly higher percentage of Japanese patients had a modified Mayo score in the moderate range and more Japanese patients in the placebo group had a Mayo endoscopic subscore of severe disease than the overall population. Japanese patients were less likely to have limited left-sided disease (Table 1). Importantly, the proportion of patients who entered the study having failed previous biologic and or tofacitinib therapies was similar to that in the overall population. Compared with the overall population, Japanese patients had lower use of prior vedolizumab. The remaining baseline characteristics were generally comparable between the Japanese cohort and the overall population. Baseline demographics and disease characteristics of the mirikizumab induction responders (maintenance population) in the Japanese subpopulation are shown in Supplementary Table 3.
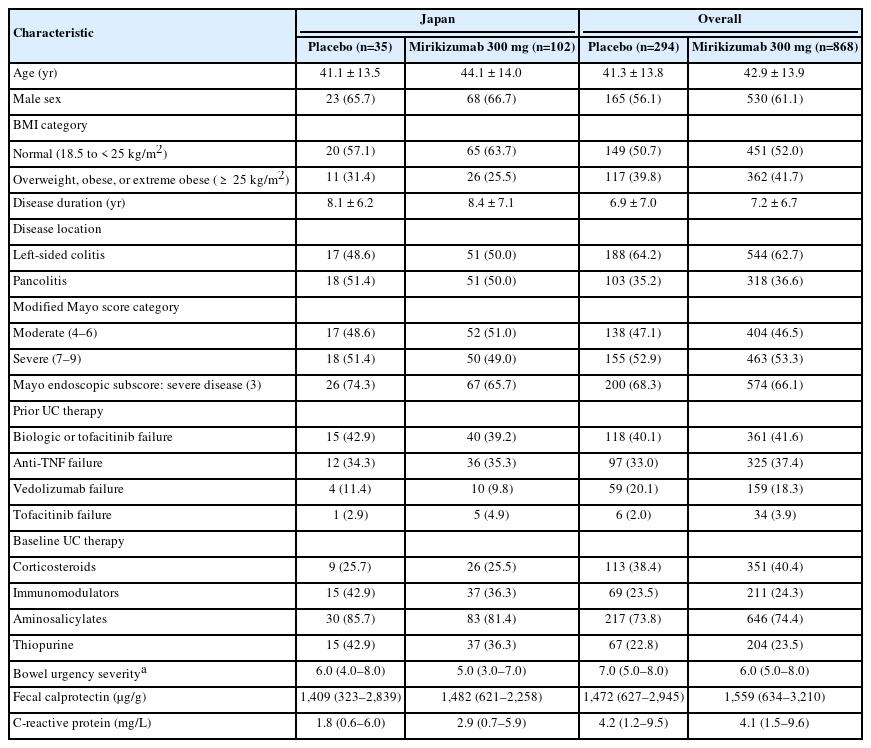
Patient Demographics and Clinical Characteristics of the Japanese Subpopulation and the Overall Population at Induction
2. Induction Therapy
At week 12 of LUCENT-1, the proportion of Japanese patients with clinical remission was numerically greater in the mirikizumab group compared with placebo (32.4% [n=33] vs. 2.9% [n=1]) (Fig. 2A); results were similar for the alternative definition of clinical remission (34.3% vs. 5.7%). In addition, clinical response, endoscopic improvement, and histologic-endoscopic improvement were numerically higher in the mirikizumab treatment arm compared with placebo (Fig. 2A). Symptomatic remission percentages over time were numerically greater in the mirikizumab group compared to placebo (Fig. 2B), and improvements in bowel urgency NRS were observed in the mirikizumab group compared to placebo (Fig. 2D), both of which are consistent with the overall population (Fig. 2C and E).
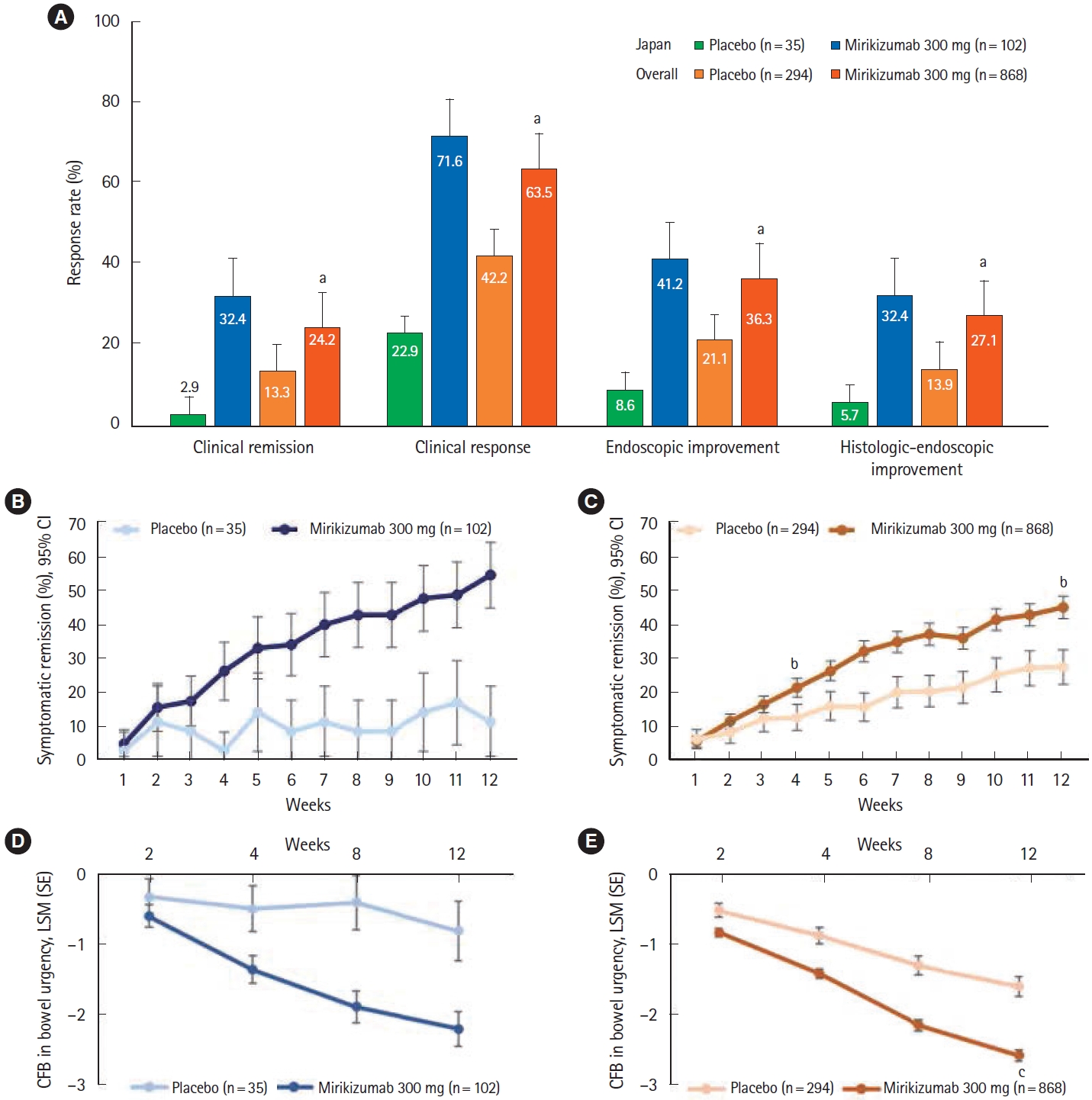
Primary endpoint and key clinical outcomes in the LUCENT-1 induction study (overall and Japanese subpopulation). The primary efficacy endpoint and key clinical outcomes in the induction study in the Japanese subpopulation was similar to the overall study population (A). In the Japanese subpopulation, symptomatic remission percentages over time were numerically greater in the mirikizumab group compared to placebo (B), and improvements in bowel urgency NRS were observed in the mirikizumab group compared to placebo (D), both of which are consistent with the overall population (C, E). aP<0.001 versus placebo, bP<0.001 versus placebo, cP<0.01 versus placebo. CFB, change from baseline; CI, confidence interval; LSM, least square means; SE, standard error; NRS, numeric rating scale.
Findings in the Japanese subpopulation in the induction study were consistent with those of the overall population where a significantly higher proportion of patients in the mirikizumab group achieved clinical remission (P<0.001) as well as a clinical response, endoscopic improvement, and histologicendoscopic mucosal improvement (all P<0.001) compared with placebo (Fig. 2). In addition, the proportion of all patients with symptomatic remission was significantly greater in the mirikizumab group at week 4 (Fig. 2C). Across the endpoints, responses in the placebo group in the Japanese subpopulation were lower compared with those in the placebo group in the overall population.
3. Maintenance Therapy
Of the 72 Japanese patients who achieved a clinical response to mirikizumab and entered the maintenance study (LUCENT-2), 47 were re-randomized to mirikizumab 200 mg and 25 were re-randomized to placebo. At week 40 of LUCENT-2 (52 weeks of treatment), the proportion of Japanese patients in clinical remission was numerically greater in the mirikizumab group compared with the placebo group (48.9% [n=23] vs. 28.0% [n=7]) (Fig. 3A); results were similar for the alternative definition of clinical remission (51.1% vs. 28.0%). In addition, the proportions of patients with corticosteroid-free clinical remission, maintenance of clinical remission, endoscopic improvement, histologic-endoscopic mucosal improvement plus absence of neutrophils, and bowel urgency (NRS of 0 or 1) were numerically greater in the mirikizumab group compared with placebo (Fig. 3A). The improvement in bowel urgency from baseline that was achieved in the induction period, as measured by the change from baseline in bowel urgency NRS, was relatively stable from week 4 to week 40 in the mirikizumab-treatment arm and numerically greater compared with placebo (Fig. 3B).
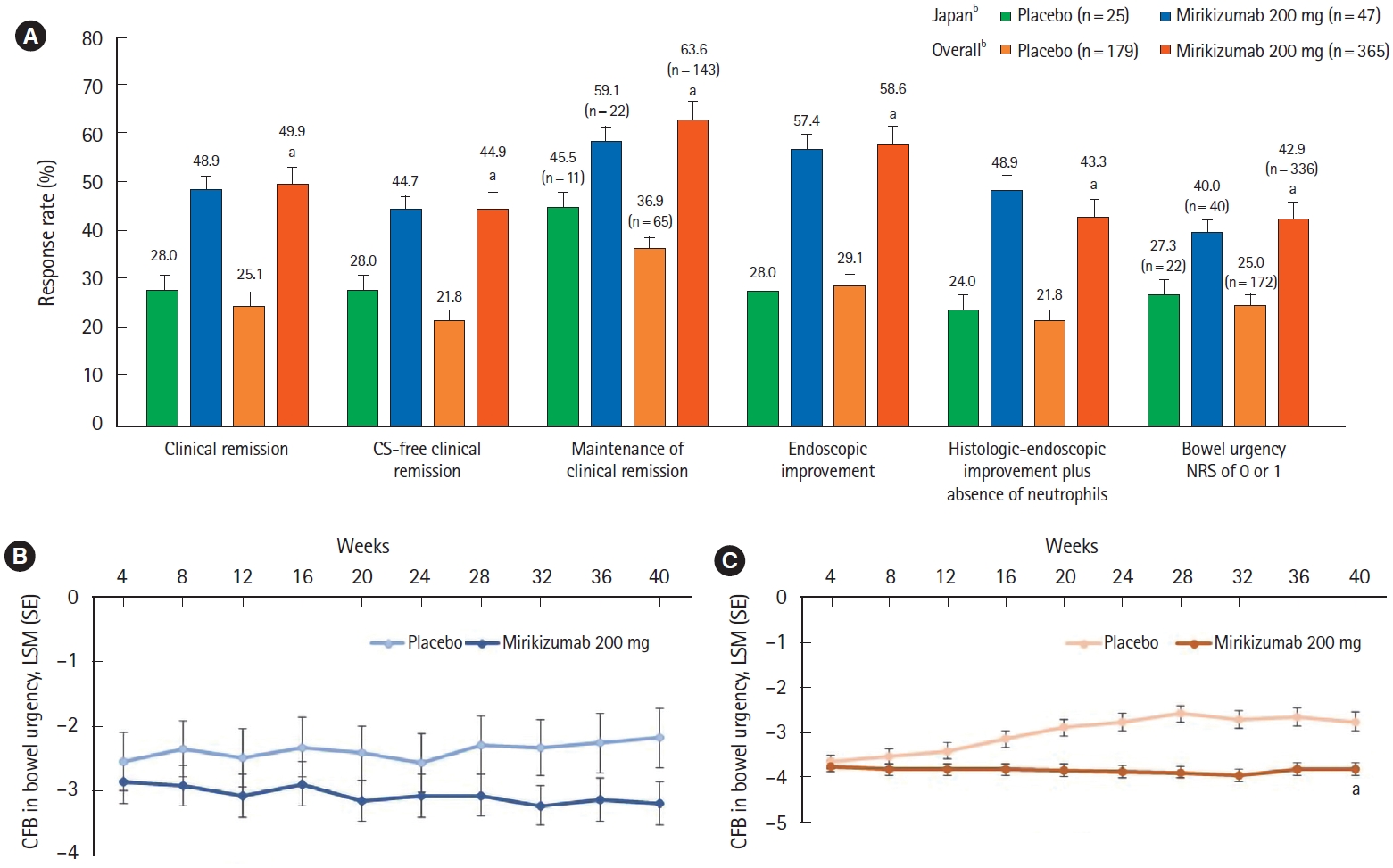
Primary endpoint and key secondary outcomes for the LUCENT-2 maintenance study (overall and Japanese subpopulation). The primary efficacy endpoint and key clinical outcomes in the maintenance study in the Japanese subpopulation was similar to the overall study population (A). In the Japanese subpopulation, the CFB in bowel urgency NRS was relatively stable from week 4 to week 40 in the mirikizumab-treatment arm and numerically greater compared with placebo (B). Statistically significant differences in the CFB in bowel urgency NRS were observed at week 40 in the overall population between the mirikizumab- and placebo-treatment arms (C). aP<0.001 versus placebo; bNumber of patients in the Japanese subpopulation and the overall population unless indicated. Maintenance of clinical remission is defined only among those achieving clinical remission by the end of induction. The percentage of patients achieving urgency NRS score (0 or 1) was calculated using patients with a baseline urgency NRS score of 3 or higher as a denominator. CFB, change from baseline; NRS, numeric rating scale; CS, corticosteroid; LSM, least square means; SE, standard error.
Overall, findings in the Japanese subpopulation in the maintenance study were consistent with those of the overall population. In the overall population, 49.9% of patients in the mirikizumab group and 25.1% of patients in the placebo group were in clinical remission at week 40 (P<0.001) (Fig. 3A). The gated secondary endpoints of corticosteroid-free clinical remission, maintenance of clinical remission, endoscopic improvement, histologic-endoscopic mucosal improvement plus absence of neutrophils, and bowel urgency (NRS of 0 or 1) were all achieved (mirikizumab group compared to placebo: P<0.001 for all endpoints) (Fig. 3A). The improvements from baseline in bowel urgency as measured by the urgency NRS remained stable throughout the maintenance period (Fig. 3C). With the Japanese population representing 11.8% of the overall population, the comparison of the “Japanese versus overall population” is comparable to that of the “Japanese versus non-Japanese” population (Supplementary Table 4).
4. Safety
The proportion of Japanese patients with TEAEs was similar between the mirikizumab and placebo groups during both the induction and maintenance studies. The incidence of TEAEs was similar between the Japanese subpopulation and the overall population in the induction study (Table 2) but was higher in Japanese patients compared to the overall population in the maintenance study for both the mirikizumab (89.4% [n=42] vs. 64.5% [n=251]) and placebo (88.0% [n=22] vs. 68.8% [n=132]) groups, attributable to the higher incidence of nasopharyngitis in the Japanese subpopulation (Table 3). The proportion of Japanese patients with serious AEs was low and comparable to that in the overall population; in both the overall and Japanese subpopulation the proportion of patients who reported a serious AE was higher in the placebo arm compared with the mirikizumab arm. There was one death reported in the placebo arm in the overall population during maintenance, which was due to COVID-19 infection.
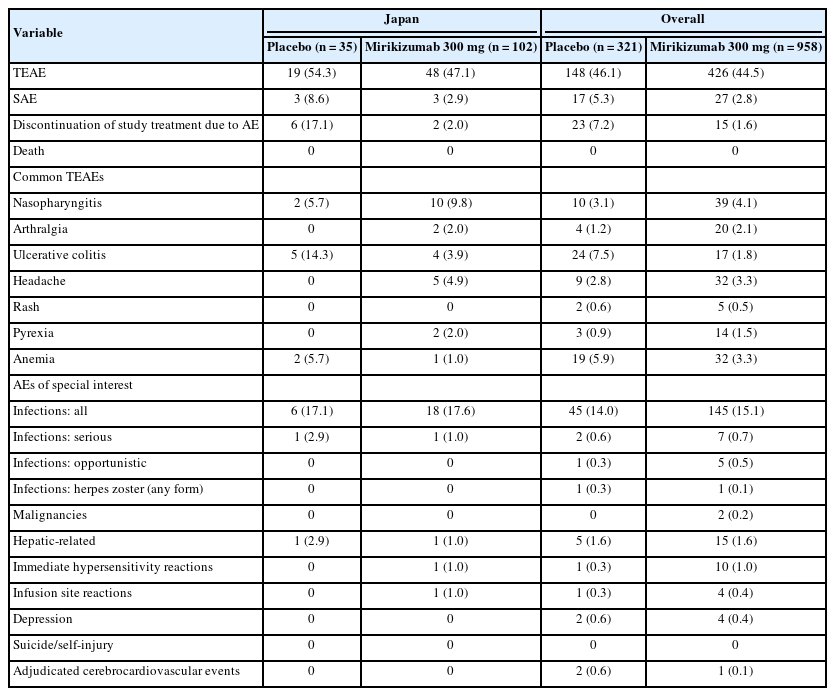
Safety Outcomes During Induction (LUCENT-1) in the Japanese Subpopulation and the Overall Population
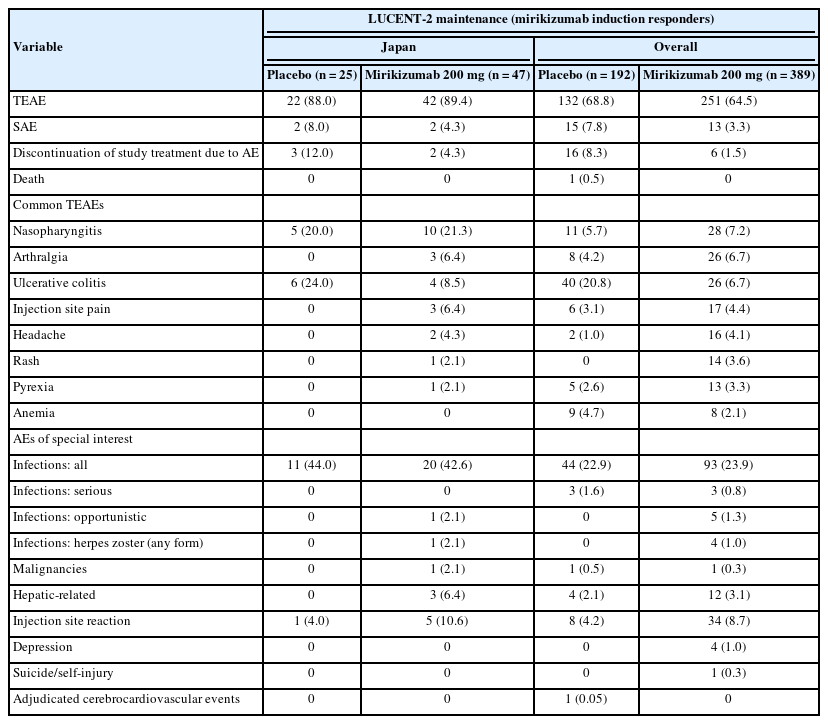
Safety Outcomes During Maintenance (LUCENT-2) in the Japanese Subpopulation and the Overall Population
In the Japanese subpopulation, the percentage of patients who discontinued study treatment due to AEs was numerically lower in the mirikizumab group compared to placebo in both trials (Tables 2, 3). In the induction trial, AEs leading to study drug discontinuation included UC in both treatment arms (placebo n=4, mirikizumab n=2), and sinusitis (n=1) and arthritis (n=1) in the placebo arm. In the maintenance trial, 3 patients in the placebo arm discontinued study treatment due to UC, and 2 patients in the mirikizumab arm discontinued due to injection site hypersensitivity (n=1) and gastric cancer (n=1).
In the Japanese subpopulation, the most commonly reported AEs during mirikizumab treatment were nasopharyngitis, UC, arthralgia, headache, and injection site pain (maintenance only). This is broadly consistent with the overall population where the most common TEAEs reported during mirikizumab treatment were nasopharyngitis, headache, and arthralgia during induction; and nasopharyngitis, arthralgia, injection site pain, headache, rash, and pyrexia during maintenance. In the mirikizumab group during induction, the proportion of the Japanese versus the overall population with nasopharyngitis was 9.8% (n=10) versus 4.1% (n=39), arthralgia 2.0% (n=2) versus 2.1% (n=20), UC 3.9% (n=4) versus 1.8% (n=17), and headache 4.9% (n=5) versus 3.3% (n=32); and during maintenance, the proportion with nasopharyngitis was 21.3% (n=10) versus 7.2% (n=28), arthralgia 6.4% (n=3) versus 6.7% (n=26), UC 8.5% (n=4) versus 6.7% (n=26), injection site pain 6.4% (n=3) versus 4.4% (n=17), and headache 4.3% (n=2) versus 4.1% (n=16) (Tables 2, 3).
Overall infections occurred with similar frequency in Japanese patients in the mirikizumab and placebo groups during both induction and maintenance. During induction, the incidence of overall infections was comparable with that of the overall population, but Japanese patients had a higher incidence during maintenance in both the mirikizumab and placebo groups. The incidences of serious and opportunistic infections were low across both studies. In the overall population, herpes zoster was the most common opportunistic infection in mirikizumab patients, however, herpes zoster was not common in the Japanese subgroup (Tables 2, 3). In both induction and maintenance studies, the severity of all infections was predominantly mild. In the Japanese cohort, there was 1 case of a severe infection (pneumonia) in the mirikizumabtreatment arm during induction. There were no alterations to the mirikizumab dose, and the patient recovered from pneumonia and continued the study. The study investigator did not consider this event related to mirikizumab treatment.
Among the AEs of special interest, there was a small numerical difference in mirikizumab-treated Japanese patients versus placebo in injection site reactions (10.6% vs. 4.0%) and hepatic-related AEs (6.4% vs. 0%) during the maintenance study. The hepatic-related AEs included increased aspartate aminotransferase (n=1), increased alanine aminotransferase (n=1), increased blood bilirubin (n=1), increased gamma-glutamyl transferase (n=1), and hepatic steatosis (n=1). All cases were considered mild and were resolved, except the case of hepatic steatosis. There was one malignancy (gastric cancer) reported in the mirikizumab group during maintenance.
DISCUSSION
This subgroup analysis of LUCENT-1 and LUCENT-2 demonstrated the efficacy and safety of mirikizumab induction and maintenance therapy in Japanese patients with moderately to severely active UC. The proportions of Japanese patients treated with mirikizumab who achieved clinical remission, clinical response, and endoscopic and histologic-endoscopic improvement during the 12-week induction trial were numerically higher than those who received placebo. Consistent with the overall population, early improvements in symptomatic remission in patients treated with mirikizumab compared with placebo were demonstrated.
Mirikizumab’s efficacy in reducing bowel urgency is particularly encouraging given a recent noninterventional study reported that UC had a significant impact on daily life in 43.5% of Japanese patients who experienced bowel urgency [12]. Overall, the proportion of participants who stated that UC “significantly impacted” their daily life was highest in participants who experienced bowel urgency or bowel incontinence [12]. Hence improvements in bowel urgency and bowel incontinence are likely to provide significant benefits in quality of life to a significant proportion of Japanese patients with UC.
Patients who achieved a clinical response to mirikizumab treatment at the end of the induction study could receive double-blind treatment with either mirikizumab 200 mg or placebo during the maintenance study. In the 40-week maintenance trial (total of 52 weeks of treatment), mirikizumab treatment demonstrated maintenance of efficacy as higher proportions of patients treated with mirikizumab achieved clinical remission, corticosteroid-free clinical remission, maintenance of clinical remission, endoscopic and histologic-endoscopic mucosal improvement plus absence of neutrophils, and bowel urgency (NRS of 0 or 1, which indicates normal or near normalization of bowel urgency) compared to placebo. Numerical improvements from baseline in bowel urgency with mirikizumab treatment were observed in the induction phase and maintained through the 40-week maintenance period in comparison to placebo. These findings in Japanese patients with moderately to severely active UC are encouraging. While clinical response and clinical remission are immediate and intermediate treatment targets for UC, respectively, endoscopic healing is a long-term treatment goal as recommended in the Selecting Therapeutic Targets in Inflammatory Bowel Disease II guidelines [13]. Although patients in the overall population who did not achieve a response to induction dosing (or lost response during maintenance) had numerically lower mirikizumab exposures than patients who achieved response (or did not lose response), due to confounding, it cannot be concluded that these lower exposures caused reduced efficacy or loss of response.
Of note, the histologic improvement component of the maintenance histologic-endoscopic mucosal improvement endpoint required the absence of mucosal neutrophils. This endpoint has been associated with improved long-term outcomes such as reduced incidence of clinical relapse and hospitalization, colectomy, and glucocorticoid use [14,15]. Following the maintenance study, approximately 49% of Japanese patients and 43% of the overall population treated with mirikizumab had absence of mucosal neutrophils in colonic biopsies.
Despite the relatively small number of patients in the Japanese subpopulation, the primary and major secondary efficacy outcomes in both the induction and maintenance studies were consistent with the findings in the overall study population [11]. This consistency is important considering the differences in the genetic background of these populations in terms of the prevalence of susceptibility genes [16]. In the global LUCENT-1 and LUCENT-2 studies, a significantly greater proportion of patients treated with mirikizumab achieved clinical remission during induction (P=0.00006) and maintenance (P<0.001) [11]. Furthermore, in both the global LUCENT-1 and LUCENT-2 studies, mirikizumab was superior to placebo in all major secondary endpoints including clinical response, endoscopic improvement, histologic-endoscopic improvement, and corticosteroid-free clinical remission (all P<0.0001), as well as significant improvements in symptomatic remission and bowel urgency [11]. In the induction phase, the response in the placebo group was lower in the Japanese subpopulation than the overall population. The reasons for this are unclear but may be due to a higher proportion of Japanese patients in the placebo group with a Mayo endoscopic subscore of 3 (severe disease) or longer disease duration.
Mirikizumab treatment during induction and maintenance was well tolerated in Japanese patients. The frequency of TEAEs was similar across mirikizumab and placebo groups in both the induction and maintenance studies. The incidence of serious AEs and study-treatment discontinuations due to AEs was low, and numerically higher in the placebo group compared with the mirikizumab group. The frequency of infections was similar between the mirikizumab and placebo treatment arms. The incidence of serious infections was very low in the induction study and no serious infections were reported in the maintenance study. The safety profile of mirikizumab observed in the Japanese subpopulation was broadly consistent with that observed in the overall population [11], and in prior studies of mirikizumab in UC [9,10], Crohn’s disease [17], and psoriasis [18]. In the maintenance study, there was a higher frequency of TEAEs (including AEs of special interest) in the Japanese subpopulation than the overall population in both the mirikizumab and placebo groups. This may be due in part to the higher incidence of nasopharyngitis in the Japanese population. The frequency of malignancies in the Japanese subpopulation was comparable to the overall population. Overall, no new safety concerns were identified in Japanese patients.
The main limitation of this subgroup analysis is the relatively small number of Japanese patients. Furthermore, the 12-week induction and 40-week maintenance periods may limit the detection and interpretation of AEs with low incidence (such as malignancy). The data from long-term extension study (LUCENT-3, NCT03519945) will provide further clarification regarding the long-term efficacy and safety profile of mirikizumab in Japanese patients with UC.
In conclusion, mirikizumab provided clinical benefits and was well tolerated in Japanese patients with moderately to severely active UC. Our findings from this subpopulation analysis of Japanese patients are in alignment with the efficacy and safety findings observed in the overall population. These findings, together with those of the overall population, support the potential of mirikizumab as a promising treatment option in Japanese patients with moderately to severely active UC where, despite advances in available treatments, a substantial unmet need persists.
Notes
Funding Source
This work was supported by the Eli Lilly and Company.
Conflict of Interest
Kobayashi T received lecture fees from Mitsubishi Tanabe Pharma, Eisai, Kyorin Pharmaceutical, AbbVie, Janssen, JIMRO, EA Pharma, Astellas, Mochida Pharmaceutical, Takeda Pharmaceutical, Gilead Sciences, Celltrion, Nippon Kayaku, and Thermo Fisher Diagnostics; advisory and/or consultancy fees from Gilead Sciences, Janssen, Pfizer, Kyorin Pharmaceutical, Mochida Pharmaceutical, Takeda Pharmaceutical, Eli Lilly and Company, Ferring Pharmaceuticals, Nippon Kayaku, Alfresa Pharma, and Bristol-Myers Squibb; and research grants from EA Pharma, Thermo Fisher Diagnostics, Alfresa Pharma, and Nippon Kayaku. Matsuoka K received research grant from Janssen Pharmaceutical K.K.; scholarship grants from AbbVie, EA, Mitsubishi Tanabe, Mochida, and Nippon Kayaku; and honoraria from AbbVie, EA, Janssen Pharmaceutical K.K., Mitsubishi Tanabe, Mochida, Pfizer, and Takeda. Watanabe M received grants from Alfresa Pharma Corporation, JIMRO Co., Ltd., Kyorin Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., and Miyarisan Pharmaceutical Co., Ltd.; grants and personal fees from AbbVie GK, Astellas Pharma Inc., EA Pharma Co., Ltd., Kissei Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., Nippon Kayaku Co., Ltd., Pfizer Japan Inc., Takeda Pharmaceutical Co., Ltd., and Zeria Pharmaceutical Co., Ltd.; and personal fees from Celgene K.K., Celltrion Healthcare Co., Ltd., Eli Lilly Japan K.K., Gilead Sciences, Inc., and Janssen Pharmaceutical K.K. Hisamatsu T has received lecture fees from AbbVie, EA Pharma, Janssen, Kyorin, Mitsubishi Tanabe Pharma, Mochida, Gilead Sciences, Inc., and Takeda, and has received scholarship donations from AbbVie, Daiichi Sankyo, EA Pharma, JIMRO, Kyorin, Mitsubishi Tanabe Pharma, Mochida, Nippon Kayaku, Otsuka, Pfizer Inc., and Takeda. Hirai F received lecture fees from AbbVie GK, EA Pharma Co., Ltd., Janssen Pharmaceutical K.K., Mochida Pharmaceutical Co., Ltd., and Mitsubishi Tanabe Pharma Co. Milch C was an employee of Eli Lilly and Company during the conduct of this study and is a former employee of Eli Lilly and Company. Hibi T received lecture fees from Mitsubishi Tanabe Pharma, Kyorin Pharmaceutical, AbbVie GK, Janssen, Mochida Pharmaceutical, JIMRO, Takeda Pharmaceutical, Gilead Sciences, Miyarisan Pharmaceutical, Kissei Pharmaceutical, Zeria Pharmaceutical, Ferring Pharmaceutical, and Nippon Kayaku; advisory/consultancy fees from Mitsubishi Tanabe Pharma, Takeda Pharmaceutical, EA Pharma, Janssen, Eli Lilly and Company, Pfizer, and Zeria Pharmaceutical; and research grants from AbbVie GK, JIMRO, and Zeria Pharmaceutical. Ishizuka T was an employee of Eli Lilly Japan K.K. during the conduct of this study and is a former employee of Eli Lilly Japan K.K. Matsuo K and Satoi Y are full-time employees of Eli Lilly Japan K.K. Milata J, Li X, Morris N, and Arora V are full-time employees of Eli Lilly and Company. Hibi T, Matsuoka K, and Watanabe M are editorial board members of Intestinal Research but were not involved in the peer reviewer selection, evaluation, or decision process of this article.
Data Availability Statement
Eli Lilly and Company provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
Author Contributions
Conceptualization: Kobayashi T, Matsuoka K, Watanabe M, Hisamatsu T, Hirai F, Hibi T, Milata J, Milch C, Ishizuka T, Matsuo K, Satoi Y. Methodology: Milata J, Milch C, Li X, Morris N, Arora V, Satoi Y. Formal analysis: Milata J, Milch C, Li X, Satoi Y. Funding acquisition: Milata J, Ishizuka T. Project administration: Milata J. Visualization: Ishizuka T. Writing-review and editing: all authors. Approval of final manuscript: all authors.
Additional Contributions
We would like to thank the clinical trial participants and their caregivers, investigators, and site staff, without whom this work would not be possible. Project management was provided by Naotsugu Akashi of Eli Lilly Japan K.K. Medical writing and editorial assistance was provided by Lisa Cossens and Antonia Baldo of Syneos Health and funded by Eli Lilly Japan K.K.
Supplementary Material
Supplementary materials are available at the Intestinal Research website (https://www.irjournal.org).
Supplemental Methods: Study Design
Average Serum Concentration of Mirikizumab in Responders and Non-responders in LUCENT-1 and LUCENT-2 (Overall Population)
Primary and Major Secondary Outcomes and Definitions for LUCENT-1 and LUCENT-2
Baseline Demographics and Disease Characteristics of the Mirikizumab Induction Responders (Maintenance Population) in the Japanese Subpopulation
Primary Efficacy Endpoint and Key Clinical Outcomes in LUCENT-1 and LUCENT-2 for the Japanese and Non-Japanese Populations
Study design of LUCENT-1 and LUCENT-2. Responders to induction mirikizumab therapy at week 12 of LUCENT-1, defined as achieving ≥2-point and ≥30% decrease in the modified Mayo score from baseline with rectal bleeding score=0 or 1, or ≥1-point decrease from baseline, were randomized to receive maintenance mirikizumab therapy or placebo in LUCENT-2. Non-responders to induction mirikizumab therapy in LUCENT-1 received additional induction mirikizumab therapy for the first 12 weeks of LUCENT-2, followed by maintenance mirikizumab therapy to week 40 in patients who achieved delayed clinical response at LUCENT-2 week 12. All patients who were expected to receive clinical benefit from ongoing access to mirikizumab (by investigator opinion) could roll over into the long-term extension study LUCENT-3. Blue boxes indicate the treatment arms used for efficacy and safety analyses in this manuscript. IV, intravenous; Miri, mirikizumab; NR, non-responder; PBO, placebo; Q4W, every 4 weeks; R, responder; SC, subcutaneous; W, week.