LIPOPOLYSACCHARIDE
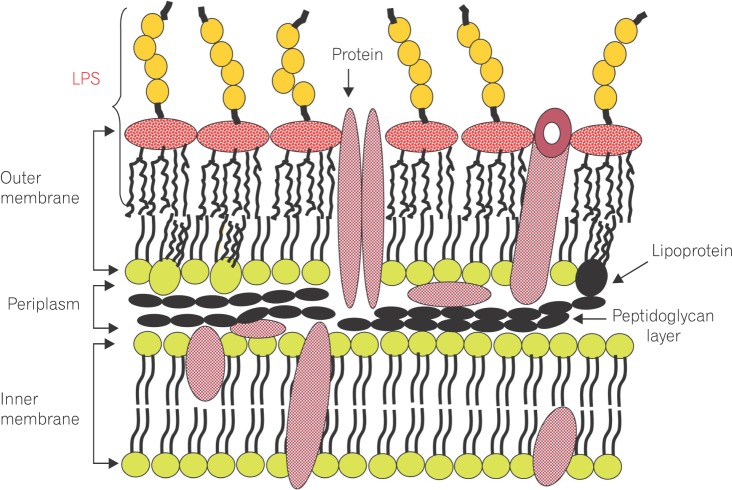
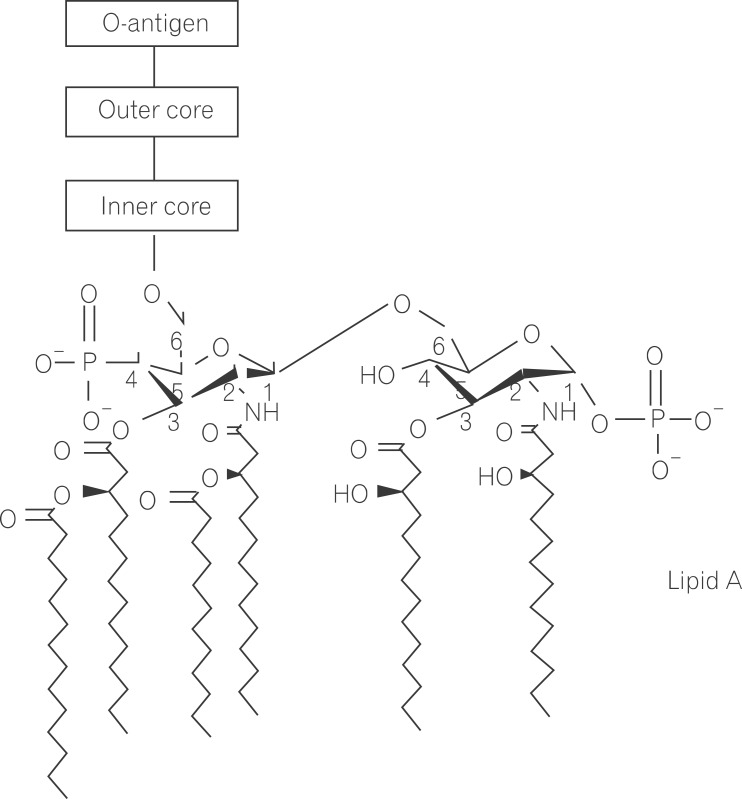
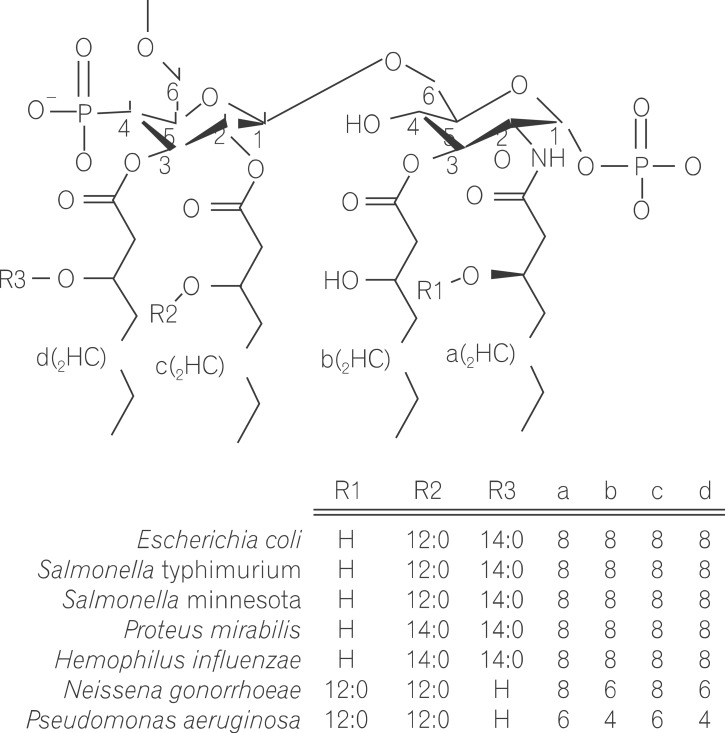
Lipopolysaccharide (LPS) is a major component in the outer monolayer of the outer membrane of most Gram-negative bacteria so as to work like a tight shield (Fig. 1). It consists of unique molecules, such as lipid A, core oligosaccharide, and O-specific oligosaccharide chain (Fig. 2).1 Lipid A is a β, 1-6 linked disaccharide of glucosamine which is acylated with R-3-hydroxymyristate at positions 2, 3, 2', and 3', and phosphorylated at positions 1 and 4'. The two R-3-hydroxy-acyl groups of non-reducing glucosamine are further esterified with laurate and myristate. However, various molecular species of lipid A are found in pathogenic gram negatives (Fig. 3). This region gives rise to biological responses that LPS induces.1,2 It is noteworthy that lipid A with six lipid chains executes full functional activity, whereas those with five lipid chains have ~100 fold less activity, and those with four lose agonistic activity. Core region of LPS is divided into the inner core and the outer core. The former part is linked to the lipid and the latter is connected to the O-specific chain. The 6' position of lipid A is glycosylated with eight-carbon sugar, 3-deoxy-D-manno-octulosonic acid (KDO). Found in most endotoxins, KDO is believed to be a potential target of therapy against LPS-associated diseases. Other core sugars include L-glycero-D-manno-heptose, glucose, galactose, and N-acetylglucosamine.2,3 The O-specific chain generally consists of 20-40 repeating saccharides, but the composition is usually different for each bacterial species. This variable region elicits production of different antibodies. Although these three segments give rise to highly complex LPS structure, lipid A is sufficient to evoke the signaling events of LPS. The entire lipid component of LPS molecule, however, is required for optimal activity. With this in mind, the individual elements appear to combine into a three-dimensional shape to facilitate interaction with the host cells.2,3
COMPLEX FORMATION OF LPS-LBP AND CD14
LPS itself is not intrinsically harmful. Instead, it acts by inducing myeloid and/or non-myeloid cells to produce a number of proinflammatory cytokines, such as tumor necrosis factor, interleukins, inducible nitric oxide synthase, and cyclooxygenase-2, which lead to fever, inadequate organ perfusion, multi-organ failure, and death observed in septic shock.4,5,6 With this in mind, LPS is a potent activator of the inflammatory responses and even minute amounts of LPS in the blood by bacterial infection are sufficient to induce potent inflammatory responses.
LPS is recognized by the core receptor complex composed of LPS-binding protein (LBP), CD14, Toll-like receptor 4 (TLR4), and MD-2. LBP is 60 KDa glycoprotein that binds to lipid A of LPS, leading to LPS-LBP complex.7 CD14 acts as a membrane receptor for LPS-LBP complex, and binding of LPS to CD14 is enhanced by LBP. CD14 is a 55 KDa protein in myeloid cells such as monocytes/macrophages. There are two types of CD14, a soluble form (sCD14) and a membrane bound form (mCD14) which is a glycosylphosphatidylinositol-anchored membrane protein, but sCD14 lacks the glycosylphosphatidylinositol-anchor.8 mCD14, however, does not have transmembrane domain to transduce LPS signaling from extracellular environment to cytosol. MD-2 is an extracellular protein responsible for LPS binding and associated with the extracellular domain of TLR4. Transfer of LPS from mCD14 to TLR4-MD-2 complex induces multimerization of the receptor complex, resulting LBP/CD14/TLR4/MD-2 complex in the plasma membrane. Subsequently, LPS bound receptor complex triggers an intracellular signaling cascade. So far, ten members (TLR1 to TLR10) of the TLR family recognizing a wide variety of microbial products have been identified in humans. Among various TLR members is TLR4 which specifically recognizes LPS and mediates LPS-induced intracellular signaling pathways.
GENETIC REGULATION OF LPS RESPONSE
Decreased sensitivity to the proinflammatory and lethal effects of LPS were observed in the C3H/HeJ mouse strain.9 C3H/HeJ mice were naturally resistant to LPS preparations from Salmonella typhosa 0-901 and Escherichia coli 0127:B8, tolerating 20 and 38 times the medial lethal dose, respectively, for other C3H sublines that are normally responsive to LPS. In addition, LPS or Lipid A is not cytotoxic in vitro for macrophages from C3H/HeJ, but directly cytotoxic for those from LPS-responsive mice.10 It was also suggested that B lymphocytes from C3H/HeJ mice were specifically hyporesponsive to LPS, but responded normally to other B-cell stimulators.11 LPS responses were also defective in T lymphocytes and fibroblasts from C3H/HeJ mice.12 These results indicated that C3H/HeJ mice were hyporesponsive to LPS. Genetically, it was demonstrated that these LPS hyporesponsive mouse strains have a defective Lps allele (Lpsd) in chromosomal assignment of the Lps locus on chromosome 4, while a normal Lps allele (Lpsn) is characteristic of most inbred mouse strains that are LPS responsive.13 Two other LPS hyporesponsive mouse strains have been identified, C57BL/10ScCr and its progenitor C57BL/10ScN (also known as C57BL/10ScNCr). It was identified that the LPS hyporesponsive phenotype of C57BL/10ScCr was due to a mutation at the Lps locus, and crosses between C57BL/10ScCr (Lpsd) and C57BL/10Sn (Lpsn) have yielded only the high-LPS responder phenotype, indicating that this Lps mutation is recessive.14
Using several techniques including cDNA selection, genomic sequencing or randomly-subcloned DNA, and comparative mapping, a candidate for Lps gene product was identified and assigned as TLR4.15,16 It was also determined that LPS hyporesponsive C57BL/10ScCr and C57BL/10ScNCr mouse strains do not transcribe TLR4 due to a chromosomal deletion in Lps gene, resulting in LPS tolerance.16 The mouse TLR4-encoding gene contains one open reading frame of 2,505 nucleotides, predicted to encode a protein of 835 amino acids. This protein consists of an extracellular domain formed by a tandem arrangement of 22 leucine-rich repeat motifs connected by a single transmembrane domain to an intracellular signaling domain that shares homology with the cytosolic region of interleukin-1 receptor (IL-1R). Furthermore, nucleotide sequencing of the entire coding region of TLR4-encoding gene in endotoxintolerant C3H/HeJ mice revealed a single missense mutation consisting of a C to A transversion at nucleotide 2,135, resulting in a substitution of proline for histidine at codon 712 (Tlr4 [P712H]) within the signaling domain of TLR4. This missense mutation within the cytoplasmic domain of TLR4 averts LPS-induced signaling, rendering the mouse non-responsive to LPS.6,15 These findings explained how C3H/HeJ mice are hyporesponsive to LPS. Identification of distinct independent mutations of the same gene in these strains of endotoxin-tolerant mice provides compelling evidence that TLR4 encoded by Lps gene is the receptor to LPS.15,16
SIGNAL TRANSDUCTION OF TLRs
TLR proteins are ligand-stimulated receptor molecules that elicit their pleiotropic effects through activation of the transcription factors nuclear factor kappaB (NFκB) and activator protein-1 (AP-1). Given that the cytoplasmic domain of TLR proteins is homologous to that of IL-1R, these regions are referred to as Toll/IL-1R (TIR) domain. Thus, the investigation of the cellular mechanism from TLR activation was approached on the basis of understanding IL-1R signaling pathway. When microbial products encounter their specific TLRs, TLRs recruit a single or a combination of adaptor molecule(s) containing TIR domain to its cytoplasmic TIR domain. At least four adaptors such as myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like/TIR domain-containing adaptor protein (Mal/TIRAP), TIR domain-containing adaptor-inducing IFN-β (TRIF), and TRIF-related adaptor molecule (TRAM) have been identified in TLR-mediated signaling pathways. After binding to the cytoplasmic TIR domain of TLRs, these adaptor molecules associate with IL-1R-associated kinases (IRAKs) to mediate the signaling to a member of tumor necrosis factor receptor-associated factor (TRAF) family (e.g. TRAF6). Thereby, TLR-induced signaling leads to activation of IKKs and MAPKs to activate transcription factors such as NFκB, AP-1, and interferon-regulatory factors, followed by inducing a pleiotropic gene expression involved in immune and inflammatory responses.17
SIGNALING CASCADE INDUCED BY LPS-TLR4 ENGAGEMENT
LPS interacts with LBP to form LPS-LBP complex which binds to CD14. However, CD14 can not mediate LPS signaling through the cell membrane, because CD14 does not have a transmembrane domain. Thus, it is believed that CD14 functions as the co-receptor for TLR4.18 Furthermore, LPS/LBP/CD14 complex interacts with TLR4. LPS responses mediated through TLR4 require the presence of MD2, a cell surface protein. All these components form the complex of LPS/LBP/CD14/MD2/TLR4.19 Upon the interaction of LPS with TLR4 and associated receptor components, TLR4 exploits the combination of Mal/TIRAP and MyD88 at the plasma membrane to induce inflammatory gene expression (called MyD88-dependent pathway). In parallel, TLR4 is internalized to endosome by the interaction with TRAM, followed by a subsequent interaction with TRIF, leading to the transcription factor activation of interferon-regulatory factors which is responsible for type-I interferon (Interferon-α/β) production (termed MyD88-independent or TRIF-dependent pathway).20,21,22 MyD88-dependent pathways are generally involved in regulating inflammatory responses and innate immunity and subsequent adaptive immunity, while MyD88-independent pathways are assigned to anti-viral activity due to the involvement of Interferon-α/β production. Given that TLR4 activation induces the signaling pathways involved in inflammatory responses, innate and adaptive immunity, and anti-viral activity, engagement of TLR4 with LPS broadly impacts a wide range of human diseases including inflammatory diseases to infectious diseases.
INNATE IMMUNITY AND BIOLOGICAL SIGNIFICANCE OF TLRs
The activation of TLRs induces the innate immunity which is an earlier evolutionary form of host defense and serves the secondary immune system by stimulating and orienting the adaptive immune responses. Indeed, mammalian TLR activation leads to expression and release of a plethora of cytokines, such as interleukins. These cytokines play direct or indirect role in adaptive immune system. It is clear that disruptions in innate immunity predispose human to microbial infections as described in the following examples. In the severely burned patient, the disruption of skin as not merely a barrier, but an organ adorned with antimicrobial peptides and first-line effecter cells like macrophages, poses great risks to microbial infections. In patients with cystic fibrosis, the alterations in salinity of the bronchial airway fluid appear to disable the function of antimicrobial peptides that are found in the respiratory epithelium, thereby leading to colonization and infection with organisms like Staphyloccoci and Pseudomonas.23,24
In the Drosophila , TOLL (Drosophila ortholog of mammalian TLR) participates in dorsal-ventral development. But in adult flies, TOLL induces the antifungal peptide, drosomycin, which is mediated through the activation of a transcription factor Dorsal.25 In comparison with TOLL, 18-wheeler causes the expression of the anti-bacterial peptide, attacin, through the activation of Dorsal-related immunity factor (DIF). While Dorsal and DIF are Drosophila NFκB proteins, Dorsal is selectively activated by TOLL and DIF is specifically activated by18-wheeler.25,26
Hundreds of anti-bacterial peptides have been reported to participate in innate immunity, not only of insects, but also of all multi-cellular organisms including human and plants. Among a wide variety of anti-microbial peptide, defensins are the representative with a wide spectra of anti-bacterial activity directed against various bacteria, fungi, and enveloped viruses.27 Although the molecular mechanisms are not fully understood, they may involve the transient appearance of channel-like structures. Defensins and most other anti-microbial peptides act by permeabilizing the cell membranes of microorganisms, resulting in the efflux of solutes. Defensins are major constituents of the microbicidal granules of blood granulocytes and are also abundantly expressed in intestinal epithelial cells specialized for host defense functions. A constitutively expressed human epithelial α-defensin is abundant in the kidney and the urogenital tract, and an infection- or cytokine-inducible β-defensin is abundant in the skin. In addition to defensins, mammals produce cathelicidins which are a group of myeloid antimicrobial peptides.28
In addition, the complement cascade plays one of the major roles in mammalian innate immunity. It is activated either directly or indirectly by microorganisms and leads to the phagocytosis through opsonization or the assembly of a pore-forming membrane attack complex on the surface of microorganism.29
IMPACTS OF LPS-TLR4 ENGAGEMENT IN THE GASTROINTESTINAL TRACT
LPS is the major constituent of the outer membrane of Gram-negative bacteria and specifically stimulates TLR4, resulting in the production of pleiotropic cytokines/chemokines. Given that the human colon harbors a large collection of commensal bacteria, the colonic lumen functions as a reservoir of LPS released from Gram-negative bacteria therein. Indeed, approximately 50 µg/mL of LPS could be observed in the human colon.30,31
The composition of intestinal microbes varies among individuals, depending on individual life conditions such as life stages, regimen, environment, genetic factors, and etc. Nevertheless, a well-orchestrated balance between Gram-negative and Gram-positive bacteria is maintained in the healthy human gut with a predominance of Gram-positive bacteria.32 Gram-negative bacteria, however, are suggested to be considerably increased in IBD patients33,34,35 and accumulated at high concentrations in an inflamed lesion of the intestine.36 Increased Gram-negative bacteria in the gut are able to increase the luminal amount of LPS, which consequently makes a strong effect on the development and perpetuation of intestinal inflammation. In line with this, our recent study demonstrated that increased LPS level in the colon resulted in the intestinal inflammation.37 Furthermore, it is worth noting that the expression of TLR4 was suggested to be increased in IBD patients.38 As a confirmatory study of this observation, the treatment of TLR4 antagonist was able to ameliorate mouse experimental colitis.39 Meanwhile, LPS was also suggested to work as a critical pathogenic factor inducing necrotizing enterocolitis.30 Together, these studies suggested that TLR4 activation by LPS should be an important pathogenic element related to the development and perpetuation of intestinal inflammatory diseases.
On the other hand, LPS was suggested to play a protective effect in a mouse epithelial injury model.40 Furthermore, LPS normally present in the gut is believed to be harmless, because the normal intestinal epithelium constantly exposed with gut microbiota might be tolerant to LPS.41,42 Although beneficial effects of gut LPS were suggested using animal experimental models, a growing body of evidence indicates that LPS in the intestine has a potency to promote the intestinal inflammation.