EPIDEMIOLOGY AND DEFINITION
Inflammatory bowel disease (IBD) is a chronic inflammatory condition affecting over 6.8 million individuals globally, impacts more than 1 million Americans and 2.5 million Europeans; it is more common among women in Western countries and men in Eastern populations and multiple studies have shown a higher incidence and susceptibility to IBD among African American, Hispanic, and Asian patients compared to Caucasians [1,2]. Several medical interventions and procedures, including immunomodulators, colectomies, and fecal microbiota transplantation, can potentially contribute to the onset or exacerbation of IBD [3]. IBD, classified as ulcerative colitis (UC) and Crohn’s disease (CD), is characterized predominantly by recurrent abdominal pain, diarrhea, rectal bleeding, severe internal cramps/muscle spasms in the region of the pelvis, and weight loss. The CD is well known for transmural inflammation and segmental involvement of the intestines, typically affecting the terminal ileum and presenting epithelioid granulomas on histological examination. Conversely, UC manifests a diffuse pattern of mucosal inflammation, primarily impacting the rectum, with variable progression toward the terminal ileum [4]. However, IBD is generally perceived as a wasting disease that can cause weight loss and malabsorption, involving various organ systems, with several extraintestinal features, including the hepatobiliary system, with findings suggesting that IBD patients have a higher prevalence of nonalcoholic fatty liver disease (NAFLD) in comparison to the general population [5]. NAFLD encompasses a spectrum of liver disorders ranging from the benign nonalcoholic fatty liver to the more severe nonalcoholic steatohepatitis (NASH), distinguished by features such as steatosis, hepatocellular ballooning, and lobular inflammation, which may progress to cirrhosis [6,7]. By early 2020, a group of international experts employed a consensus-driven process, utilizing a 2-stage Delphi consensus to introduce the term “metabolic dysfunction-associated fatty liver disease (MAFLD)” aimed to bring resolution to a prolonged debate regarding the renaming of NAFLD to align the term more accurately with the disease process and solidify its classification as a metabolic disorder (Table 1).
With increasing urbanization, westernization, and the increasing issues of obesity, diabetes, and hypertension, the occurrence of MAFLD escalated from 16% in 1988 to 37% in 2018 in the United States. Simultaneously, the worldwide prevalence surged from 15% and 33% in 2005 to 25% and 59.1% in 2010. Currently, the global prevalence of MAFLD stands at 50.7% among overweight or obese adults, with males (59.0%) showing a notably higher prevalence compared to females (47.5%) [8-10]. MAFLD is often distinguished with patients exhibiting hepatic steatosis confirmed through histology, imaging, blood markers, or visible fat accumulation alongside meeting one of the following criteria: being overweight or obese (according to ethnicityspecific thresholds), having type 2 diabetes mellitus, or displaying indications of metabolic dysregulation. The latter is defined as having 2 or more of the following conditions: (1) an increased waist circumference; (2) high blood pressure or taking specific medications; (3) elevated triglyceride (TG) levels or taking specific medications; (4) low levels of high-density lipoprotein cholesterol; (5) prediabetes; (6) a high homeostatic model assessment of insulin resistance score; or (7) inflammation as indicated by elevated levels of high-sensitivity C-reactive protein [11]. Over the past decade, notable advancements have been made in comprehending the relationship between NAFLD and IBD, as these patients are more prone to develop NAFLD and liver fibrosis despite the absence of traditional risk factors [12]. This review aims to study the association between NAFLD and IBD and its impact on patient care, as the coexistence of these 2 clinical entities might pose a therapeutic challenge.
PATHOPHYSIOLOGY
IBD is typically thought of as a condition that causes significant weight loss and malabsorption, often resulting in malnourishment. Several studies have indicated that individuals suffering from IBD often have protein and energy malnutrition with loss of muscle mass (sarcopenia), an independent risk factor for NAFLD in patients with IBD. A diet with a calorie deficit of at least 9% of the total energy requirement or a protein and amino acids deficiency often increases the TG accumulation in the liver, resulting in steatosis and severe inflammation [13]. In addition, insufficient protein in the diet also lowers peroxisomes. It affects mitochondrial function adversely, increasing lipid content inside the hepatocytes. Hepatic peroxisomes and mitochondria play a significant role in the metabolism of nutrients by providing substrates for aerobic metabolism and lipogenesis by early regulation of fatty acid metabolism [14].
1. Malnutrition and Vitamin Deficiency
Malnourishment in patients suffering from IBD often predisposes them to micronutrient deficiencies like vitamin D deficiency, folate deficiency, and vitamin A deficiency, which often results in NAFLD [15-17]. Recent clinical evidence suggests that vitamin D and hepatic vitamin D receptors (VDRs) may play a role in the pathogenesis of NAFLD. It has been pointed out that liver VDR expression can modulate intrahepatic lipid accumulation, possibly by regulating the provincial levels of angiopoietin-like protein 3 and lipoprotein lipase. The vitamin D/VDR axis increases the expression of the insulin-dependent glucose transporter 4 on adipocytes and upregulates the intracellular expression of IRS-1, the insulin receptor substrate, which in turn promotes glucose uptake in muscle cells. These metabolic pathways are linked to insulin sensitivity and glucose-insulin homeostasis. Furthermore, vitamin D deficiency decreases the insulin secretory response to glucose loading, and VDR transcriptionally regulates the insulin gene in pancreatic β cells [15,18]. Furthermore, vitamin D deficiency often results in defective tight junctions, resulting in increased translocation of lipopolysaccharide into the blood, hence predisposing to the vicious cycle of IBD and NAFLD. Moreover, the vitamin D/VDR axis modulates gut microbiota, which affects the development of NAFLD in obesity [16,17]. In addition to vitamin D deficiency, vitamin A and folate are often implicated in the pathogenesis of NAFLD in patients of IBD as they play an essential role in glucose and lipid metabolism in the liver and adipose tissue [19].
1) Vitamin A
Like vitamin D, vitamin A deficiency has been proposed in the pathogenesis of NAFLD, which is usually deficient in patients with CD. Vitamin A metabolites, particularly retinoic acids, play a role in regulating these processes directly. The primary control of hepatic de novo lipogenesis (DNL) is through the transcriptional regulation of the sterol response element binding protein-1c (SREBP-1c) and carbohydrate response element binding protein (ChREBP). These proteins induce the expression of critical enzymes involved in glycolysis (glucokinase, pyruvate kinase isozymes R/L, ATP citrate lyase, acetyl-CoA synthetase) and lipid synthesis (acetyl-CoA carboxylase 1, fatty acid synthase, ELOVL fatty acid elongase 6, stearoyl-CoA desaturase-1, glycerol-3-phosphate acyltransferase [GPAT], mitochondrial) in the liver. The inhibition of SREBP-1c or ChREBP reduces hepatic steatosis by impairing lipid synthesis. Insulin, glucose, and fructose stimulate the expression of SREBP-1c and ChREBP, promoting hepatic DNL [16,20,21].
Retinoic acids and synthetic ligands of retinoid X receptor (RXR)α, such as bexarotene, are known to enhance hepatic DNL and plasma TG levels by activating liver X receptor (LXR)/RXR and peroxisome proliferator-activated receptor γ (PPARγ)/RXR. This, in turn, enhances the expression of SREBP-1c and ChREBP. LXR and PPARγ are typically activated by oxy cholesterols and non-esterified fatty acids, respectively, with RXRα being a permissive dimerization partner as RXR ligands enhance DNL through those heterodimers independently of the co-presence of ligands for LXR or PPARγ. In addition, LXR/RXR also directly induces fatty acid synthase expression, promoting DNL and enhancing plasma TG levels in mice [22-24]. In contrast, all-trans retinoic acid (atRA) suppresses DNL by activating retinoic acid receptor α (RARα). This, via induction of Hes family BHLH transcription factor 6 (HES6) and subsequent inhibition of hepatocyte nuclear factor 4α (HNF4α), reduces PPARγ expression and downstream SREBP-1c activity. In a counteracting mechanism, 9-cis retinoic acid-activated RXRα induces expression of a small heterodimer partner (SHP), which inhibits HES6 expression, promoting DNL via the PPARγ-SREBP-1c axis. However, SHP also inhibits LXR/RXR transcriptional activity, thereby simultaneously inhibiting SREBP-1c-mediated DNL. This highlights the delicate position of SHP in the development of hepatic steatosis, inhibiting RXR/LXR-ChREBP/ SREBP-1c-mediated DNL while at the same time promoting DNL via the HES6-HNF4α-PPARγ pathway. The absence of SHP protects mice from diet-induced hepatic steatosis, suggesting a most prominent role for the HES6-HNF4α-PPARγ axis, which atRA and RARα activate. Hepatic TG synthesis is catalyzed by GPAT, mannosyl (α1,6-)-glycoprotein beta-1,2-N-acetylglucosaminyltransferase, and diacylglycerol O-acyltransferase 2, all of which are under the transcriptional control of ChREBP. In addition, GPAT is controlled by SREBP-1c. Although no specific data are available on the RA-mediated expression of these genes, they are likely coregulated with essential genes in DNL due to RXR/LXR and RAR-mediated effects on ChREBP and SREBP-1c [16,25,26].
In addition, the hepatic uptake of TG-containing chylomicron remnants or low-density lipoprotein (LDL) particles is regulated by the LDL receptor (LDLR). The expression of human LDLR is controlled by the LXR and RXR. However, the co-regulation of LDLR by retinoic acids is yet to be studied in detail. Based on the potent effects of RXR ligands on LXR/RXR-mediated regulation of SREBP-1c and ChREBP, it is likely that retinoic acids may also promote the LDLR-mediated uptake of TGs in the liver. Hepatic very LDL (VLDL) particle formation and secretion are facilitated by apolipoprotein CIII (apo-CIII). A genetic variant of apo-CIII leads to enhanced circulating apo-CIII in humans and is associated with NAFLD [27,28]. RARα suppresses hepatic apo-CIII expression through a pathway involving SHP and HNF4α, thereby reducing hepatic and plasma TG levels. Earlier studies have shown that RXR ligands have the opposite effect and enhance hepatic apo-CIII expression, either via RXR homodimers or RXR/PPARα, thereby promoting hypertriglyceridemia. Hypertriglyceridemia is a wellknown adverse effect of pharmacological ligands of RXR and a risk factor for cardiovascular disease. This emphasizes the opposite roles of RAR ligands (such as 9-cis retinoic acid) and RXR ligands (such as 9cRA) on VLDL particle production and secretion by the liver [16,27,28].
In addition, vitamin A modulates the lipolysis of TGs inside the hepatocytes through a complex mechanism involving the adipose TG lipase (ATGL/PNPLA2), hormone-sensitive lipase, and PNPLA3 through the effects of PPARα and PPARα agonist. Liver diseases, especially those that lead to hepatic fibrosis, are often linked to disturbed vitamin A homeostasis. Liver injury triggers hepatic stellate cells (HSCs) to transdifferentiate into myofibroblasts, which produce excessive amounts of extracellular matrix, leading to fibrosis. HSCs lose their retinyl ester stores during this process, leading to vitamin A deficiency [26]. Additionally, PNPLA3, a hepatic enzyme, often acts as a TG hydrolase, an acetyl-CoA-independent transacylase, and a retinyl esterase. PNPLA3 was initially thought to have hydrolase activity towards TGs. Specifically, those containing mono- and poly-unsaturated fatty acids with various genotypes found on different locus and associated with NAFLD-associated genetic risk factors, particularly PNPLA3-I148M, which also predisposes for disease progression and NAFLD-associated hepatocellular carcinoma. The PNPLA3-I148M variant reduces hydrolase activity. It causes fat accumulation in liver cells, a primary characteristic of NAFLD. Recent studies have shown that PNPLA3 is present in HSC and has retinyl esterase activity, promoting the release of retinol from lipid droplets. Individuals carrying the PNPLA3-I148M allele with NAFLD or obesity alone have reduced fasting circulating retinol and retinol binding protein 4 levels, but no association was found with β-carotene. Hepatic retinyl palmitate levels are increased in individuals homozygous for PNPLA3-I148M, suggesting a role for PNPLA3 in hepatic retinoid metabolism. PNPLA3-I148M variant promotes fibrogenic features of HSC, including enhanced proliferation, migration, and expression of collagen type 1 alpha 1, pro-inflammatory cytokines, and chemokines alongside lower cellular retinol levels. PNPLA3-I148M-carrying HSC contains more lipid droplets, a typical characteristic of HSC quiescence. These features increase the risk for progressive liver disease in PNPLA3-I148M carriers. However, which hepatic cell type retinyl esters accumulate in NAFLD, specifically in PNPLA3-I148M patients, is unclear [16,29-31].
2) Folate
Like vitamin A and vitamin D, patients suffering from IBD are also deficient in other micronutrients, like folate, which is involved in the pathogenesis of NAFLD. Folate, involved in one-carbon metabolism, has been implicated in NAFLD and NAFLD-related comorbidities [16]. Numerous studies have shown that folate deficiency may contribute to the development of steatosis in rodents, as folate deficiency leads to high expression of lipid biosynthetic genes, which perturbs lipid metabolism in the liver. This depletion also impaired hepatic lipid transport by VLDL in mice [32]. As discussed earlier, folate is essential for synthesizing S-adenosylmethionine from methionine which is involved in an enzymatic reaction by providing a methyl donor to an enzyme phosphatidylethanolamine N-methyltransferase catalyzing the methylation of phosphatidylethanolamine (PE) to phosphatidylcholine (PC), which is necessary for VLDL assembly in hepatocytes. During folate deficiency, the synthesis of PC from PE by phosphatidylethanolamine N-methyltransferase is reduced, which may compromise lipid export by VLDL and promote hepatic lipid accumulation. A low PC-to-PE ratio may also enhance cell membrane permeability, leading to leakage of cellular components, activation of immune cells, and cytokine secretion. Moreover, PC is a significant phospholipid required for liver bile secretion. Bile production, crucial for the absorption and digestion of dietary fats, was also decreased in folate-deficient rodents. Inhibition of bile production may have adverse effects on lipid metabolism [16,32-34].
Hepatic insulin resistance can also perturb the regulation of lipid metabolism and is a common feature observed in human and rodent models of NAFLD. The glucose and lipid production regulation is disrupted when insulin action is compromised in the liver. Abnormal lipid and carbohydrate metabolism in the liver is often associated with dysregulation of AMP-activated protein kinase (AMPK) [35]. The AMPK is a crucial regulator of metabolism that corresponds with energy balance. Inactivation of AMPK has been associated with hepatic lipid accumulation, hyperglycemia, and hyperinsulinemia in animal models with high-fat diet-induced NAFLD. A recent study demonstrated that folic acid supplementation restored AMPK activation in high-fat diet-fed mice, improving hyperinsulinemia and lipid and glucose metabolism. Therefore, the hepatoprotective effect of folate may be attributed to its essential role in metabolic regulation [16,36].
In addition to one-carbon metabolism, folate acts as a scavenger for reactive oxygen species, regulates the activity of reactive oxygen species-generating enzymes, and restores the activity of antioxidant enzymes. Folate can also reduce the production of inflammatory cytokines secreted by immune cells [37]. Studies indicate that folic acid supplementation can improve lipid metabolism and oxidative stress in rodent models of NAFLD. However, there are concerns about folic acid over supplementation, which can mask vitamin B12 deficiency and impair cognitive function. To minimize adverse effects, periodic trials are necessary to determine the optimal dose of folic acid supplementation in specific populations, such as individuals with fatty liver disease [16].
2. Gut Microbiota Dysfunction/ Intestinal Dysbiosis
IBD often shows changes in gut microbiota, which leads to NAFLD. The altered microbiota damages the tight junctions between cells, allowing bacterial endotoxins like lipopolysaccharides to be translocated into the bloodstream. These endotoxins activate Toll-like receptors and trigger downstream signaling cascades that induce the transcription of inhibitor of nuclear factor kappa B to activate nuclear factor kappa light-chain-enhancer of activated B cells, intensifying lipids’ oxidation in the liver and resulting in NAFLD [38]. In addition, gut microbiota’s choline, PC, and l-carnitine metabolism produces trimethylamine (TMA). TMA is then further oxidized into the metabolite TMA-N-oxide, which plays a crucial role in the pathogenesis of NAFLD. Also, the gut microbiota plays a vital role in the synthesis of bile acids [39]. Intestinal dysbiosis often results in decreased bile salt hydrolase production, leading to a lower synthesis of bile acids, causing an increased risk of cholesterol and TG accumulation, and predisposing to dyslipidemia [40,41]. In addition, patients with IBD often have increased intestinal permeability, leading to improper immune reactivity against the saccharomyces cerevisiae antigen with a resultant increase in anti-Saccharomyces cerevisiae antibodies with perinuclear anti-neutrophil cytoplasmic antibodies is necessary serodiagnostic tests in patients with IBD. Increased intestinal permeability often increased the prevalence of small bowel bacterial overgrowth in these patients, resulting in the alteration of gut microbiota, which could be a pathogenic link between IBD and NAFLD [19,42].
3. Metabolic Syndrome
Studies have demonstrated that metabolic syndrome (MS), in addition to malnourishment, is a risk factor for NAFLD in individuals with IBD [43,44]. The mechanism behind the development of NAFLD in IBD patients with MS is similar to that of non-IBD patients, involving multiple insults through insulin resistance, nutritional factors, hormones secreted from adipose tissue, and genetic and epigenetic factors [45]. Insulin resistance leads to the release of specific chemokines like RANTES/CCL5, MIG/CXCL9, IL-8/CXCL, and IP-10/CXCL10, resulting in a pro-inflammatory state in the body through the migration of various inflammatory cells, including T cells, monocytes, dendritic cells, and neutrophils, and attaching to transmembrane receptors like CCR1, CCR3, and CCR5, predisposing to MS and hence, to hepatic steatosis and fibrosis often resulting in NAFLD [46,47]. In addition to increased insulin resistance levels, a low choline intake often found in obese populations resulting in its deficiency often predisposes to the development of NAFLD as it decreases the LDL output from hepatocytes, increasing the fat accumulation inside the hepatocytes. In addition, there is a strong correlation between body mass index, choline consumption, and the degree of steatosis, as shown by a study in which obese individuals who increased their choline intake from 272 mg/day to 356 mg/day showed a reduction in the fatty liver index, particularly those with a higher body mass index (mean = 35.9 kg/m2). Therefore, choline supplementation may help modulate fatty liver index in obese people with NAFLD [48,49].
4. IBD Therapeutics
1) Glucocorticoids
Glucocorticoids (GC) are often used to manage patients suffering from IBD. However, some patients with poorly controlled diseases may require repeated or prolonged exposure to GCs, significantly affecting carbohydrate and lipid metabolism and leading to an increased risk of developing MS and potentially NAFLD [50].
GCs facilitate adipose tissue hyperplasia by increasing the transformation of preadipocytes into adipocytes [51]. Elevated levels of GCs inside the body often impair glucose uptake in skeletal muscle and adipose tissue, leading to hyperglycemia and subsequent hyperinsulinemia and insulin resistance inside the body. Hyperglycemia and hyperinsulinemia, combined with insulin resistance, often pose a risk for steatosis inside the hepatic parenchyma. In addition to the hyperglycemia and peripheral insulin resistance, treatment with GCs can mimic Cushing disease, which is associated with elevated cortisol levels and insulin resistance not only in peripheral tissues but also inside the hepatic tissue, which increases the risk for MS and hence development of NAFLD [19,52,53].
GCs also inhibit the β-oxidation of fatty acids by inhibiting the transcriptional activity of PPAR-α, consequently increasing lipid accumulation. Also, GCs increase free fatty acids released from adipocytes and increase the circulating of free fatty acids inside the blood with increased TG accumulation inside the hepatocytes by a decrease in the degradation of apolipoprotein B and reducing the activity of triacylglycerol hydrolase, thus decreasing the hydrolysis of TG inside the liver [54]. However, there is still no data on what dose and duration of exposure to GC may lead to the development of steatosis, especially in humans [19].
2) Methotrexate
Methotrexate (MTX) is a stable derivative of aminopterin, the first folic acid antagonist that inhibits DNA synthesis, repair, and cellular replication, and is used in treating various malignant and non-malignant immune-mediated disorders such as RA, psoriasis, and CD. It is often used with tumor necrosis factor (TNF)-α inhibitors in inducing remission in IBD patients [55]. MTX acts by competitively inhibiting the enzyme dihydrofolate reductase, which inhibits thymidylate and purine synthesis, decreasing the production of DNA and RNA [22]. Initially, it was thought that the primary mechanism of MTX’s anti-inflammatory role on IBD patients was through this pathway. However, later studies proved that the increased levels of adenosine and inhibition of polyamines, among others, were the primary mechanisms of the anti-inflammatory role of MTX [56]. Once absorbed, 10% of MTX is converted to 7-hydroxy MTX, which is taken up intracellularly by reduced folate carrier 1, and the 10% of MTX/7-hydroxy MTX is metabolized to MTX polyglutamate which inhibits intracellular 5-aminoimidazole4-carboxamide ribonucleotide (AICAR) transformylase. With the inhibition of AICAR transformylase, there is a subsequent increase in intracellular AICAR, inhibiting essential steps in adenosine degradation. This leads to increased adenosine accumulation in the intracellular and extracellular space. This adenosine then binds to its receptors (A1, A2a, A2b, and A3), and is responsible for significant anti-inflammatory effects, including inhibiting lymphocyte proliferation and producing cytokines such as TNF, interleukin (IL)-8, and IL-12 [55,57]. However, MTX is well-known for elevating liver transaminase levels. However, it is even more concerning for its link to changes in liver histology, especially fibrosis and cirrhosis. Patients on MTX may display histologic changes in liver biopsies such as steatosis, stellate (Ito) cell hypertrophy, and hepatic fibrosis [58]. However, the exact mechanism responsible for MTX-induced hepatotoxicity still needs to be clearly understood. Various hypotheses exist, including MTX activation of Ito cells and vitamin A-storing lipocytes in the peri-sinusoidal areas. Chronic liver injury activates them into myofibroblasts that secrete collagen and other matrix proteins, such as fibronectin. Another theory is that prolonged intracellular accumulation of MTX, particularly MTX polyglutamate, can cause prolonged folate depletion, which is required for DNA synthesis [55,59]. However, there is still no data on what dose and duration of exposure to MTX may lead to the development of steatosis, especially in humans. Moreover, currently, there are no universally accepted guidelines for monitoring MTX in patients with CD.
3) Thiopurines
Thiopurines are prodrugs converted into an active metabolite called 6-thioguanine that stops the purine metabolism of cells in constant replication, such as activated T lymphocytes. The most commonly used thiopurines in clinical practice are mercaptopurine (MP) and azathioprine [60]. These drugs are used to maintain remission in patients with IBD, either alone or in combination with anti-TNF biological therapy, to reduce the formation of antibodies and prevent the loss of effectiveness of these biologics. However, the use of thiopurines in patients with IBD carries a risk of drug-induced liver injury. In some studies, this risk has been reported to be between 3.7% and 13.3% [61]. Thiopurine involvement in the liver can manifest in various ways, from asymptomatic increases in liver tests to acute cholestatic or mixed hepatitis and vascular endothelial lesions, including sinusoidal dilatation, peliosis of the liver, sinusoidal obstruction syndrome (formerly termed Veno-occlusive disease), and regenerative nodular hyperplasia. While cholestatic hepatitis and elevated aminotransferases are idiosyncratic reactions and occur mainly within the first 3 months after starting treatment, vascular endothelial lesions are dose-dependent and present more frequently between 3 months and 3 years after starting treatment with thiopurines [62]. Several studies have highlighted the increased risk of liver damage from thiopurines and MTX in people with pre-existing liver disease who have IBD. Therefore, it is essential to consider the presence of any liver disease or hepatic steatosis before starting any immunomodulators. The development of drug-induced liver injury due to thiopurines is related to the activity of the thiopurine S-methyltransferase enzyme. This enzyme plays a crucial role in the metabolism of MP and azathioprine. Genetic polymorphisms may decrease the activity of this enzyme, leading to higher levels of 6-methyl MP metabolites, which increases the risk of hepatotoxicity [63,64].
4) Parenteral Nutrition
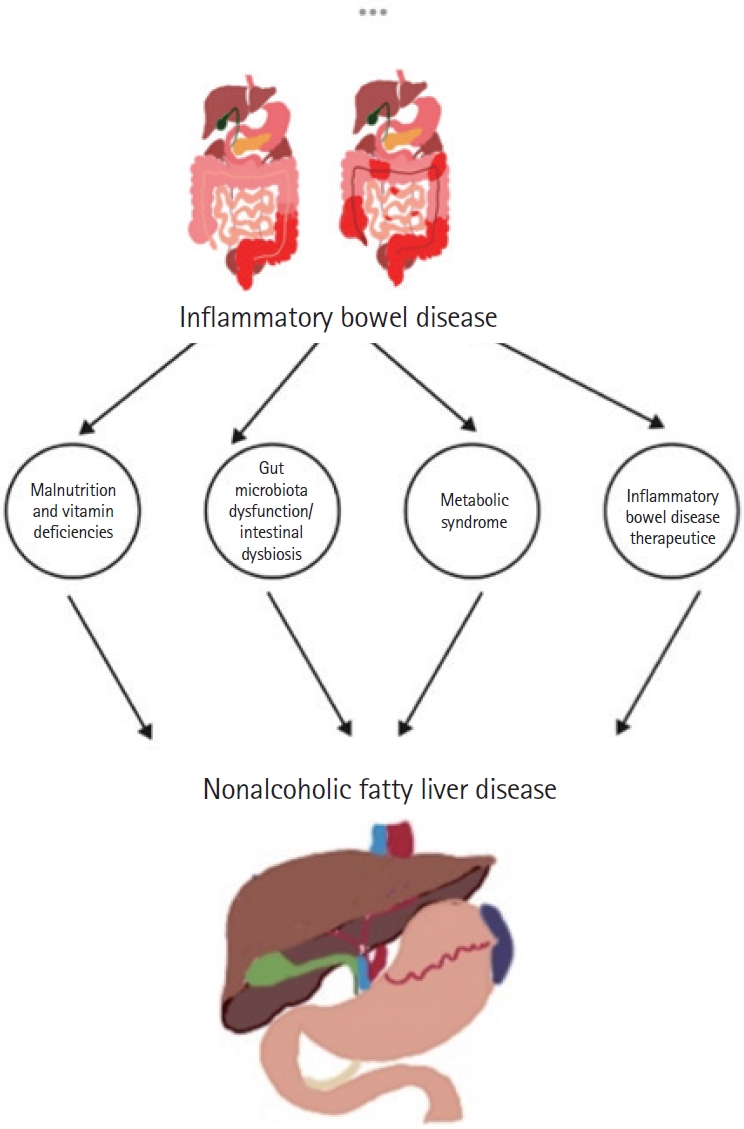
Some patients with IBD may require parenteral nutrition (PN) due to extensive surgical resection or refractory disease, resulting in intestinal failure. However, exposure to PN for 5 days may lead to hepatic steatosis and prolonged exposure to PN, eventually leading to progressive inflammatory response and fibrosis. These issues may be exacerbated by excessive caloric and carbohydrate intake. Additionally, deficiencies in amino acids such as carnitine and choline and essential fatty acids may also be contributing factors in developing NAFLD in patients with IBD (Fig. 1) [50,65].
DISCUSSION
1. Risk Factors
NAFLD is one of the most prevalent liver conditions in Western nations, and its importance as a health concern is growing [44]. Many recent studies and case reports have demonstrated the cooccurrence of NAFLD and IBD, suggesting multiple risk factors for this combination disorder (Table 2) [66,67]. Onwuzo et al. [68] have shown that in comparison to individuals without IBD, patients with IBD had a higher prevalence of developing NAFLD. According to the multivariate analysis, people with CD (odds ratio [OR], 2.79; 95% confidence interval [CI], 2.58-3.02) and UC (OR, 2.37; 95% CI, 2.17-2.60) had roughly twice as high risk of developing NAFLD than people without IBD. NAFLD was found in 48% of patients with CD and 44% of patients with UC in a retrospective study involving 694 IBD patients [69]. Obesity and dietary preferences have been seen to act as major risk factors in developing hepatic steatosis in patients with IBD. It is seen that IBD patients tend to consume more fats and proteins and avoid dietary fiber, fruits, and vegetables because they believe that these foods will cause symptoms like diarrhea and abdominal distention [5]. Overconsumption of fat-rich food, particularly from animal sources, is associated with obesity and the generation of pro-inflammatory molecules [70]. This could lead to inflammation and alterations in the microbiome and may play a role in developing hepatic steatosis [71]. Additionally, Magrì et al. [5] found a positive link between NAFLD and high serum insulin and basal glucose levels, explaining a shared pathogenic mechanism for both NAFLD and IBD. Advanced age is found to be another risk factor for NAFLD in patients IBD (OR, 1.04; 95% CI, 1.01-1.07; P=0.006), highlighting the role of age-related lipid accumulation and free radical injury and prolonged production of inflammatory cytokines due to underlying IBD [5]. Aging is also associated with a decrease in muscle mass, known as sarcopenia, which has also been found to affect the functioning of the liver and accumulation of fat. Kim et al. [72] followed 12,624 patients without underlying NAFLD in their longitudinal study over 7 years to analyze the changes in skeletal muscle mass. They found that 15% of the 12,624 patients without a baseline NAFLD developed incident NAFLD throughout their study. Several studies involving American and Italian populations have also indicated that increased skeletal muscle mass may be linked to a decreased incidence of NAFLD and possibly even the resolution of pre-existing NAFLD, indirectly supporting the relationship between sarcopenia and NAFLD [72,73]. IBD is also commonly associated with sarcopenia, with a prevalence of 37% and 52% in UC and CD, respectively. This may explain the presence of a common underlying pathogenic pathway; however, more research is needed to prove this analogy [74]. The evidence of fatty changes in the liver has also been found to be associated with small bowel surgeries like small bowel resections, which are particularly performed in patients with uncontrolled IBD. The pathogenesis of fatty changes is linked to altered fat handling in the gut with resections, which causes elevation in plasma free fatty acids, decrease in carnitine concentration, and deficiency of essential fatty acids [43]. Another crucial risk factor which may be responsible for NAFLD is the use of corticosteroids (CS) therapy for IBD. However, the role of CS is debatable as the existing literature has shown contrasting results with many studies favoring this relationship whereas many others suggest no underlying mechanism [44,75,76]. The research which supports the role of CS in NAFLD, however, highlights an indirect effect of CS involving obesity, insulin resistance and MS. Therefore, whether to restrict the use of CS or to cautiously use them is a debate for more research. Sartini et al. [77] have highlighted an important concept of NAFLD phenotypes in the setting of IBD. According to them the NAFLD in patients with underlying IBD is different from the NAFLD in patients without an underlying IBD as the severity of IBD is directly related to the amount of steatosis detected on ultrasound. They concluded that the individuals with severe IBD had a higher frequency of severe steatosis during ultrasonography (USG) in comparison to individuals with mild-to-moderate disease (32.1% vs. 16.6%, P=0.01) [70]. This is further supported by a cross-sectional study by Abenavoli et al. [69] which suggests that a longer and severe IBD course may act as a fertile ground for the risk factors of NAFLD to flourish, particularly chronic inflammation and alterations in gut microbiota. The gut dysbiosis could potentially be a major factor in the development and progression of IBD-related NAFLD.
Many recent studies have come up with the idea of disparity in the impact of UC and CD on NAFLD. Kodali et al. [78] have concluded in their systematic review that the patients with CD have higher risk of NAFLD than the patients with UC. Distinct pathophysiological pathways that underlie CD and UC may be responsible for differences in the development of NAFLD. While UC is restricted to the colon and causes surface inflammation, CD is characterized by transmural inflammation and can affect any part of the gastrointestinal system. This variation in anatomical distribution and severity of inflammation in CD, may explain a higher risk of metabolic disturbances and hence higher degree of NAFLD [78].
2. Clinical Features
Most NAFLD patients do not show symptoms, but some may report fatigue, upper right abdomen discomfort, enlarged liver (hepatomegaly), acanthosis nigricans, or increased fat deposits. Cirrhosis might be the first sign for many, with 48% to 100% of NASH cases being asymptomatic, often discovered incidentally during unrelated medical checks. Though chronic liver failure signs are rare, around 25% might have an enlarged spleen at diagnosis. Abnormal liver function tests (raised alanine aminotransferase [ALT] and aspartate aminotransferase [AST] but usually below 250 IU/L) or accidental discovery of liver fat on imaging often lead to NASH/NAFLD diagnosis. Hepatomegaly, due to fatty infiltration, can be noticed during a physical exam [79]. Histopathological analysis through liver biopsy remains the gold standard for staging the disease but due to its invasive nature, liver biopsy poses a risk of serious complications, occurring in approximately 1% of cases [80]. Patients with consistently high levels of AST and ALT in their blood and fatty changes on a USG or computed tomography scan may be considered to have NAFLD [81]. Typical results seen on USG are bright liver echo patterns, more echogenicity compared to the renal cortex, and decreased resolution of structures inside the liver [5].
3. Treatment
As of now, there is no officially approved drug preparation for treating NAFLD. Vitamin E, obeticholic acid (a farnesol X-receptor agonist), and pioglitazone (a PPAR agonist) may help improve the pathological signs of NAFLD. A study by Vilar-Gomez et al. [82] of 261 NAFLD patients who had liver biopsies taken before and after making changes to their lifestyles to lose weight found that more weight loss is linked to better histologic features of NAFLD. Patients who lost at least 10% of their body weight had the highest rates of NASH reduction (100%), NAFLD resolution (90%), and fibrosis regression (45%). In 2010, the PIVENS trial emerged as the most extensive randomized vitamin E study. It involved 247 adults with biopsy-confirmed NAFLD but without diabetes. The trial compared vitamin E (800 IU once daily), Pioglitazone (30 mg once daily), and a placebo. Their findings revealed that vitamin E treatment significantly improved NAFLD more than the placebo (43% vs. 19%, P=0.001) [83]. In a 4-month randomized placebo-controlled clinical trial by Ni et al. [84], the participants with diagnosis of NAFLD were administered resistant starch as a microbiota-directed dietary supplement. The trial yielded a significant reduction of 9.08% in intrahepatic TG content. After adjusting for weight loss, this reduction remained substantial at 5.89%. Hence administration of resistant starch through food or supplement may help in NAFLD, however, its use particularly in the patient with IBD requires substantial research [84]. Another important modality is the use of anti-TNF-α agents. In many animal models, anti-TNF-α agents have shown to decrease hepatic inflammation and steatosis. However, some animal models have also demonstrated the protective effect of TNF-α in the liver. This contrary data arises confusion with use of drugs that antagonizes the effect of TNF-α. In a recent retrospective cohort study by Tang et al. [85], no beneficial effects of anti-TNF-α agents were observed in the development of NAFLD or its progression to cirrhosis. The study included 226,555 patients who were followed for 1.5 years with anti-TNF-α agents for NAFLD with increased hazards of 1.53 (95% CI, 1.32-1.77). Another meta-analysis comprising 5 studies has come up with mixed results, even suggesting an increased risk of liver steatosis in patients using anti-TNF-α [86].
4. Clinical Course
Various studies have shown that IBD can have an impact on the development of NAFLD, but little is known about how NAFLD may affect the clinical outcomes of IBD. Preliminary reports suggest that NAFLD may worsen outcomes for people with IBD, leading to increased hospitalizations, mortality rates, lengths of stay, and healthcare costs. The impact of NAFLD on IBD outcomes needs to be better understood due to the complex relationship between these 2 conditions. Dysregulation of lipid metabolism and intestinal dysbiosis are believed to play a role in both diseases. These processes may produce various biomolecules with pathological effects, potentially leading to increased oxidative stress and promoting hepatic steatosis while also worsening IBD activity. IBD-related hospitalizations account for around two-thirds of all IBD costs, so it is essential to understand how comorbidities like NAFLD may impact the clinical outcomes of people with IBD [87,88].
5. Follow-up
Patients having NAFLD with underlying IBD need to be followed up regularly not only for managing the IBD but also the NAFLD counterpart. The goals of follow-up should be established based on current evidence and guidelines but should also be tailored according to the patient’s disease. The protocol should include early search for NAFLD during the initial diagnosis of IBD, and then followed by timely follow-up to monitor the progression of NAFLD, predict response to the treatment trials and identify individuals with worse prognosis. Studies suggest that a combination of modalities can be used for diagnosing and following up patients which includes both imaging and biochemical testing. Ultrasound is usually preferred as the first line imaging modality of choice for diagnosing NAFLD, however, a modified version known as FibroScan (a type of elastography which measures liver stiffness) can be used at regular intervals for follow-up purposes [89]. The biochemical tests include detecting specific biomarkers which are associated with the progression of disease and development of fibrosis such as hyaluronic acid, α-2-macroglobulin, tissue inhibitor of metalloproteinase 1 (TIMP1), and procollagen type III N-terminal peptide (P3NP). The various biomarker tests include the FibroTest®, the FibroMeter®, the ELF, and the HepaScore®. However, these specialized tests are less validated when it comes to their utilization in following up patients with NAFLD having an underlying IBD. Boursier et al. [90] found that the rate of excluding advanced liver disease in patients with NAFLD was 30% higher with the FibroMeter® than with non-specific FIB-4 score (which includes 4 variables including age, AST, ALT, and platelets) [90]. However, the use of such tests for follow-up and assessing the progression of hepatic steatosis to hepatic fibrosis is a topic for further research. Although there is no clear cut data available to establish the follow-up of NAFLD with underlying IBD, it is crucial to define practical rules/guidelines to avoid the repeated use of different modalities which could lead to significant costs and non-judicial use of resources. However, there is a general agreement on the use of noninvasive tests for follow-up which can be repeated every 3 years and which can be decreased to 2 years in patients having additional risk factors for disease progression such as diabetes, hypertension and MS (Fig. 2) [89].
CONCLUSION
The relationship between IBD and NAFLD demonstrates a complex interplay of numerous factors influencing the development and course of the disease. Although there is evidence between NAFLD and IBD due to established risk factors such as age, obesity, and type 2 diabetes, recent research shows the relationship between metabolic dysregulation and IBD relapses, highlighting the significance of elements such as dysbiosis, mucosal destruction, and elevated cytokines during active disease stages. In summary, NAFLD and IBD research demands a comprehensive understanding of metabolic and inflammatory interactions. Knowing the complexities of these interconnected disorders is essential for tailoring effective therapeutic strategies and providing holistic care to individuals dealing with them.