INTRODUCTION
Colorectal cancer (CRC) is one of the most common cancers worldwide. Most CRC cases (approximately 70%) are sporadic and lack significant genetic components [1]. In contrast, approximately 30% of all CRC cases are genetically predisposed [2]. Hereditary CRC is a subset of genetically predisposed CRC that occurs because of inherited genetic mutations and accounts for up to 5% of all CRC cases [3]. Lynch syndrome (LS) and familial adenomatous polyposis (FAP) account for an estimated 2%-4% and < 1% of all CRCs, respectively, and are representative hereditary CRC types characterized by mutated genes [4,5]. These gene mutations increase the risk of developing CRC at an early age and can also increase the risk of developing other types of cancer. The timely identification of individuals with hereditary CRC is crucial for implementing appropriate surveillance and prevention strategies, ultimately reducing morbidity and mortality rates.
Hereditary CRC encompasses a group of diseases or syndromes resulting from mutations in specific genes that lead to distinct clinical phenotypes (Table 1) [2,5,6]. These conditions include LS, FAP, MUTYH-associated polyposis (MAP), juvenile polyposis syndrome (JPS), Peutz-Jeghers syndrome (PJS), Cowden syndrome, and polymerase proofreading-associated polyposis.
Genetic counseling is recommended for individuals with a personal or family history of hereditary CRC syndromes [7,8]. During genetic counseling, a healthcare professional with expertise in genetics will assess an individual’s personal and family history, inform the benefits and limitations of genetic testing, and provide information about the potential implications of the test results for the individual and their family members. Genetic testing may involve the analysis of specific genes associated with hereditary CRC syndromes to identify diseasecausing mutations. Genetic tests include testing for only one (e.g., APC for FAP) or more than one (MLH1/MSH2/MSH6/PMS2/EPCAM for LS) causative gene, multigene panel tests, and whole genome sequencing (Table 2) [5,6]. Multigene panel testing (MGPT) to use next-generation sequencing (NGS) technology may be more efficient than single-gene testing in some cases, although it has a higher chance of identifying variants of uncertain significance and receiving overscreening and overtreatment [9,10]. In addition, NGS technology can detect genetic defects such as large deletions and mosaicism, which cannot be detected using direct sequencing techniques [9,11]. The identification of individuals with hereditary CRC syndromes through genetic testing allows for personalized surveillance and prevention strategies. This can involve regular colonoscopy for polyp removal, endometrial cancer screening, risk-reducing surgeries, and other measures to manage and reduce the risk of cancer development. In this review, we elucidated the screening and surveillance strategies for LS, FAP, and other hereditary CRC syndromes.
SCREENING AND SURVEILLANCE STRATEGIES
Screening and surveillance strategies play a crucial role in the early detection and treatment of CRC and related diseases [12,13]. These strategies aim to identify individuals who may have early signs or are at a high risk of developing the diseases. By detecting CRC in its early stages, treatment options may become more effective and potentially lifesaving [14].
LYNCH SYNDROME
LS is the most common hereditary CRC syndrome, accounting for approximately 2% to 4% of all CRC cases [15,16]. LS is an autosomal dominant disorder caused by germline mutations in mismatch repair (MMR) genes including MLH1, MSH2, MSH6, PMS2, and EPCAM (Table 1) [17]. LS is associated with an increased risk of colorectal, uterine, endometrial, ovarian, stomach, small bowel, and urothelial cancers [18].
1. Screening
Screening for LS is essential for the early detection and prevention of CRC and other associated cancers in at-risk individuals. Typically, screening involves a combination of clinical criteria, family history, and genetic testing.
1) Clinical Criteria
For the selection of individuals, clinical criteria are cost-effective. The Amsterdam criteria II and the revised Bethesda guidelines were used to identify individuals who should undergo further testing for hereditary non-polyposis colorectal cancer (HNPCC) (Table 3) [19,20]. These criteria include the age at which CRC is diagnosed, presence of other HNPCC-related cancers, and family history.
The Amsterdam criteria, from the International Collaborative Group on Hereditary Non-Polyposis Colon Cancer in Amsterdam, in 1990, had limited sensitivity. Thus, the Amsterdam criteria II was introduced in 1999 [20], and one study in 2000 reported a sensitivity of 78% and specificity of 61% [21,22].
The Bethesda guidelines were introduced by the National Cancer Institute in 1996 in an international workshop on HNPCC. These were revised in 2004 [19]. The revised Bethesda guidelines have been shown to be highly effective in identifying carriers of MSH2/MLH1 mutations in patients with newly diagnosed CRC. With a combination of microsatellite instability (MSI) testing, the sensitivity and specificity are 81.8% and 98.0%, respectively [23,24]. However, it is important to note that these criteria are intended as guidelines for selecting patients who require further testing and are not designed to confirm the presence of HNPCC.
2) Family History
A detailed family history helps identify patterns of cancer occurrence that may suggest HNPCC. Healthcare professionals typically look for multiple family members with CRC or other HNPCC-related cancers, as well as cases diagnosed at a young age.
3) Genetic Testing
Genetic testing plays a crucial role in HNPCC diagnosis. Genetic testing is recommended when an individual meets clinical criteria and has a family history suggestive of HNPCC. For HNPCC-related cancer, the initial step is to perform an MSI test or immunohistochemistry for MMR proteins (MLH1, MSH2, MSH6, and PMS2) in tumor tissue samples [25]. These tests help to identify cases with defective MMR function, indicating a potential underlying genetic mutation.
If MSI or immunohistochemistry tests suggest an MMR deficiency, further genetic testing is performed to identify specific mutations in MLH1, MSH2, MSH6, PMS2, or EPCAM genes. Sequencing of the individual genes involved in MMR helps to confirm the diagnosis of HNPCC and identify the specific affected genes. In recent years, MGPT has emerged as an alternative strategy for genetic testing in patients with CRC [10]. MGPT utilizes NGS technology to simultaneously analyze multiple genes associated with a particular family of cancer phenotypes or multiple phenotypes. Instead of selecting genes based on tumor characteristics or family history, the MGPT tests a predefined panel of genes, including the MMR genes, along with other cancer-related genes. The advantage of MGPT is that it allows for a comprehensive analysis of multiple genes associated with different cancer types, enabling the identification of potential hereditary cancer syndromes beyond HNPCC. However, it is important to note that the broader scope of MGPT may increase the likelihood of identifying variants of uncertain significance or incidental findings that require further evaluation and genetic counseling.
2. Surveillance
Surveillance strategies for individuals with LS aim to detect and manage early-stage cancers or precancerous lesions (Table 4) [5,6,10,26]. However, different surveillance methods are appropriate for different types of cancer.
1) Colorectal Cancer
Regular colonoscopy is the primary surveillance tool for LS. Colonoscopy allows for the detection and removal of precancerous polyps and early-stage CRC. It is usually recommended to start surveillance at the age of 20-25 years or 2-5 years earlier than the age of the youngest family member diagnosed with CRC, if it is before the age of 25 years, whichever comes first [3]. Colonoscopy should be repeated every 1 to 2 years [27]. According to British guidelines, the age at onset of surveillance colonoscopy should be stratified according to the LS-associated gene, and colonoscopy should be recommended from 25 years of age for MLH1 and MSH2 mutation carriers, and 35 years of age for MSH6 and PMS2 mutation carriers [6].
However, even with regular colonoscopy, there is still a risk of developing CRC. The Prospective Lynch Syndrome Database, the largest prospective database of known MMR carriers, has shown the development of a considerable number of CRC cases despite colonoscopy with polypectomy [8,28]. This finding suggests that colonoscopy has some limitations as a surveillance tool for LS. MLH1 and MSH2 carriers have a lifetime CRC risk of up to 50%, which remains high although of a regular colonoscopy surveillance [8]. In contrast, MSH6 and PMS2 carriers have a lower lifetime CRC risk than MLH1 and MSH2 carriers [24]. There are several hypotheses regarding the development of CRC despite undergoing regular colonoscopies, including missed lesions, rapid progression, and adenoma-free progression to cancer [24].
2) Gastric Cancer
Screening and surveillance for gastric cancer in patients with LS are essential for early detection and better clinical outcomes. However, according to British guidelines, gastric surveillance in patients with LS should only be conducted in the context of a clinical trial [6]. Although there are no consensus guidelines specifically for LS-associated gastric cancer, esophagogastroduodenoscopy (EGD) starting at the age of 30-40 years is recommended every 2 to 4 years in areas with a high prevalence of gastric cancer [10]. High-risk individuals with a family history of upper gastrointestinal cancer or high-risk endoscopic findings are recommended to undergo annual or biennial EGD [10]. According to Japanese guidelines, EGD is recommended for patients with a family history of gastric cancer starting at the age of 30-35 years and should be performed every 1 to 3 years in areas where gastric cancer is prevalent [5]. Helicobacter pylori infection is a significant risk factor for gastric cancer. Screening for H. pylori infection and subsequent eradication therapy are recommended. Screening can be performed using a breath test or biopsy during endoscopy.
3) Endometrial Cancer
Women with LS should undergo annual screening for endometrial cancer starting at the age of 30-35. This approach involves a combination of transvaginal ultrasonography and endometrial biopsy.
4) Ovarian Cancer
Transvaginal ultrasound can be used to visualize the ovaries and monitor any changes in their size or structure. Although it has not been proven to significantly reduce ovarian cancer mortality, it may be useful in high-risk individuals. As a screening method, transvaginal ultrasound is recommended at the age of 30-35 years in high-risk individuals and should be performed annually thereafter. For women at a high risk of developing ovarian cancer due to LS, risk-reducing bilateral salpingo-oophorectomy (BSO) is recommended. This procedure has been shown to significantly reduces the risk of ovarian cancer [29]. The optimal age range for considering BSO is typically 40-45 years after completion of childbearing [17,23]. The decision to undergo BSO as a risk-reducing option, as well as the optimal timing, should be individualized and carefully considered.10
5) Other Cancers
In addition to colorectal, endometrial, ovarian, stomach, and small bowel cancers, LS has been associated with an increased risk of certain other cancers. The specific recommendations for screening these cancers may vary based on family history and the specific gene mutations involved (Table 4). Recommendations for screening other LS-associated cancers are still evolving, and there may be variations in the screening protocols and guidelines. A specific screening approach should be tailored to individual risk factors, family history, and genetic mutations.
(1) Pancreatic cancer
Individuals with LS may have a higher risk of developing pancreatic cancer, especially those who are MLH1 carriers [30]. There are limited data on the pancreatic cancer risk among MSH2 and MSH6 carriers. PMS2 carriers have not been shown to be at an increased risk of pancreatic cancer [30]. Screening options may include imaging studies such as annual magnetic resonance imaging (MRI), magnetic resonance cholangiopancreatography, and endoscopic ultrasound to detect early signs of pancreatic abnormalities. According to the National Comprehensive Cancer Network (NCCN) guidelines, MLH1 carriers with at least one first- or second-degree relative with exocrine pancreatic cancer should undergo pancreatic cancer screening starting at age 50 and then annually [6]. The American College of Gastroenterology guidelines for pancreatic cancer screening state that screening should be considered for people who have a strong family history of pancreatic cancer, defined as one or more first-degree relatives with pancreatic cancer [21].
(2) Prostate cancer
LS is also associated with an increased risk of prostate cancer in male individuals. Prostate cancer screening may involve regular prostate-specific antigen blood tests and digital rectal exams as recommended by current guidelines for prostate cancer screening. The NCCN guidelines recommend consideration of prostate cancer screening at age 40 years should be considered and then annually [6]. The American College of Gastroenterology does not recommend prostate cancer screening for people with LS unless they have a family history of prostate cancer [21].
(3) Urinary tract cancer
LS is also associated with an elevated risk of urinary tract cancers, including cancers of the ureter and renal pelvis. Screening methods may involve periodic imaging studies, such as urinalysis, computed tomography (CT) scans or ultrasonography, to monitor the urinary tract for abnormalities. The NCCN guidelines state that there is no evidence to support surveillance for urinary tract cancers in LS [6]. However, the guidelines also state that surveillance may be considered in those with a family history of urinary tract cancer [6].
FAMILIAL ADENOMATOSIS POLYPOSIS
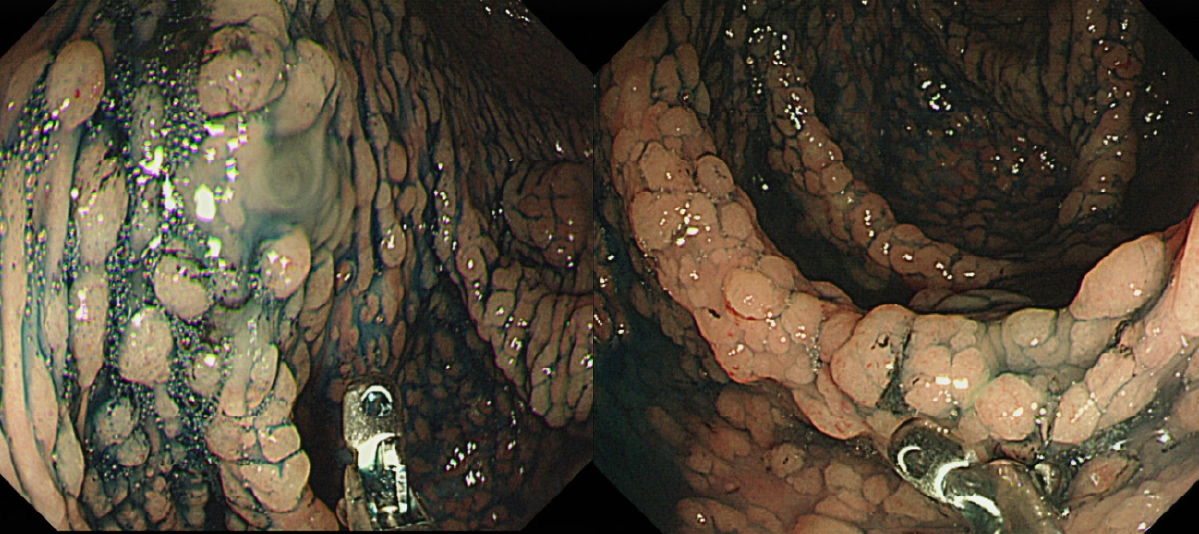
FAP is the second most common hereditary CRC syndrome, accounting for approximately 1% of all CRC cases [4,31]. FAP is an autosomal dominant disorder caused by germline mutations in the APC gene that results in the development of hundreds to thousands of adenomatous polyps in the colon and rectum (Fig. 1) [32]. These polyps can become cancerous if left untreated. Therefore, regular screening and surveillance are crucial for detecting and managing potential complications in individuals with FAP.
1. Genetic Testing
Genetic testing can confirm the presence of FAP by identifying mutations in APC gene. This gene is associated with WNT/ β-catenin signaling, and mutations in the gene can lead to the development of polyps in the colon. Genetic testing is usually performed in individuals with a family history of FAP or when clinical suspicion is high. If a person is found to have a mutation in the APC gene, they are considered as having FAP and will need to be monitored closely for the development of polyps and cancer. Genetic testing can also be used to identify at-risk family members. If a person is found to have a mutation in APC, their family members can be tested to determine whether they have inherited the mutation. This information can guide appropriate surveillance and management strategies for at-risk family members.
According to the NCCN guidelines, surveillance for FAP in families with known APC mutations can be guided by the results of an APC gene test [10]. Individuals with a positive APC mutation are at very high risk of developing CRC, and they should be monitored closely with colonoscopy every 12 months beginning at 10-15 years. Individuals that do not have the APC mutation may be screened for as having average risk, which typically involves colonoscopy every 5-10 years beginning at 45-50 years of age. However, no significant APC gene mutations were detected in approximately 10% of patients with FAP. Individuals who have not been tested for APC mutations should discuss the advantages of genetic testing with their healthcare providers. If the APC test is incomplete, individuals should be monitored for the development of polyps and cancer with careful colonoscopic surveillance. Detailed screening and surveillance recommendations for FAP patients are described in Table 5.
2. Colonoscopy
Colonoscopy is the primary method for screening and surveillance of FAP [34]. According to the American Society for Gastrointestinal Endoscopy recommends colonoscopy screening should begin at the age of 10-12 years, with annual follow-ups, for individuals at risk for FAP (Table 5) [5,6,10,26,33]. The frequency of follow-up colonoscopies may be individualized based on the number of polyps found. An attenuated phenotype ( < 100 adenomas) may not require frequent colonoscopies, unlike a classical phenotype ( > 100 adenomas) [6].
Individuals with a confirmed diagnosis of atypical FAP often develop CRC at a later age. Screening is recommended to begin in late teens to the early 20’s and colonoscopy should be performed every 1-2 years [35].
According to the NCCN guidelines, endoscopic surveillance after surgery can vary depending on the type of surgery performed [10]. Patients who have undergone colectomy with ileorectal anastomosis (IRA) should undergo endoscopy for evaluation of the rectum every 6-12 months depending on the number of polyps [10]. Patients who have undergone total proctocolectomy with ileal pouch-anal anastomosis (IPAA) should undergo annual endoscopic evaluations of the ileal pouch and rectal cuff, depending on the polyp burden [10]. The frequency of surveillance should be shortened to every 6 months for large, flat polyps with villous histology and/or high-grade dysplasia [10].
In addition to colonoscopy, imaging techniques such as virtual colonoscopy (CT colonography) or MRI may be used as adjuncts to screen for polyps in patients with FAP. These noninvasive methods provide detailed images of the colon and other FAP-related organs and can be particularly useful in cases where colonoscopy is contraindicated or not well tolerated.
3. Esophagogastroduodenoscopy
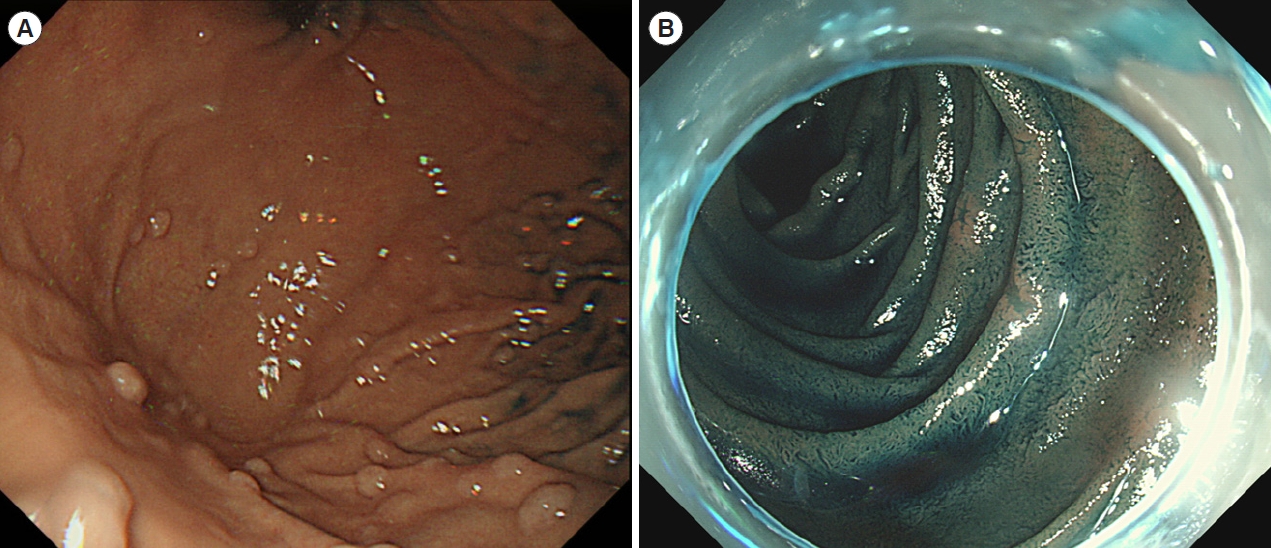
Individuals with FAP are also at risk of developing duodenal and gastric polyps as well as periampullary and duodenal adenocarcinomas (Fig. 2) [26]. The American Society for Gastrointestinal Endoscopy recommends upper gastrointestinal endoscopy surveillance starting at the age of 20-25 years for patients with FAP, with follow-up intervals based on the Spigelman stage (Tables 6 and 7) [26,36]. The risk of gastric cancer in patients with FAP appears to be higher in patients from geographical areas with a high risk of gastric cancer. Therefore, screening for H. pylori infections and subsequent eradication therapies are recommended. In addition, the ampulla of Vater should be observed during regular surveillance, and ampullary adenomas require endoscopic papillectomy (Fig. 3).
4. Desmoid Tumor
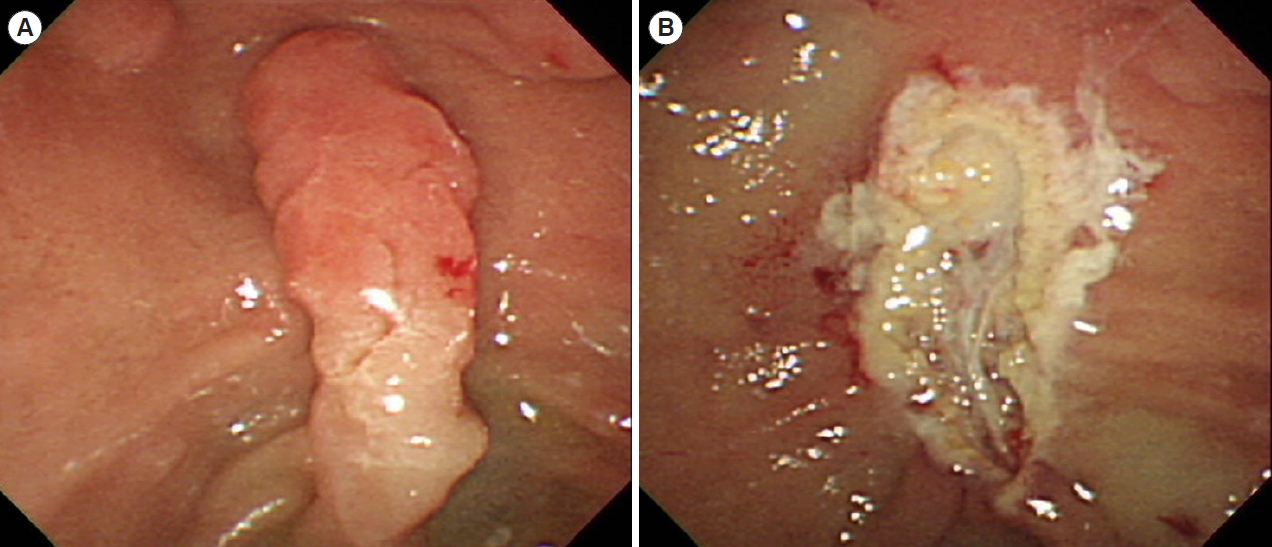
Desmoid tumors are a type of benign fibroma that can also be invasive tumors in patients with FAP [5]. They are the leading causes of death in patients with FAP, with an incidence rate of approximately 10% [5]. Specific mutation sites in the APC gene and family history may be risk factors for the development of desmoid tumors. Desmoid tumors typically develop in the abdominal wall, mesentery, or retroperitoneum, particularly after surgery (Fig. 4) [5]. They can grow very large, and infiltrate other structures causing symptoms and complications. Desmoid tumors are usually diagnosed using imaging tests such as CT or MRI. There is no cure for desmoid tumors, but there are treatments that can help control their growth, including surgery, radiation therapy, medications such as celecoxib/tamoxifen, and chemotherapy. Individuals with a history of desmoid tumors should be closely monitored for the development of new tumors. Imaging tests such as CT or MRI, should be performed every year or twice a year to detect new tumors early [37].
5. Surveillance of Other Organs
FAP is associated with an increased risk of developing various extracolonic tumors including thyroid cancer, hepatoblastoma, and brain tumors. Especially, regular surveillance of thyroid is recommended (Table 5), and the surveillance of other organs for rare diseases may vary depending on individual factors and family history.
MAP SYNDROMES
MAP is an autosomal recessive genetic condition characterized by the development of multiple adenomatous polyps in the colon and the rectum. Individuals with MAP have an increased risk of developing CRC [10].
If MAP is suspected based on clinical symptoms or family history of the condition, a diagnostic workup is typically performed. This may include a thorough medical history review, physical examination, endoscopy and imaging tests, including EGD and colonoscopy, and genetic testing to identify mutations in MUTYH.
Regular colonoscopy is the mainstay of surveillance for patients with MAP. The recommended starting age for colonoscopy varies, but screening is often recommended in the late teens, early twenties, or 5-10 years before the earliest diagnosis of CRC in the family. The frequency of colonoscopy may range from 1 to 3 years, depending on the individual’s polyp burden and other risk factors. According to the NCCN, colonoscopy is recommended every 1 to 2 years beginning at the age 25 to 30 years [10].
During colonoscopy, polyps found in the colon and rectum are removed or biopsied for pathological examination. This helps to reduce the risk of CRC. Management of detected polyps depends on their size, number, and histological characteristics. Large or advanced neoplasia may require more aggressive interventions.
If the polyp burden is high in the colon and rectum, colectomy with IRA or proctocolectomy with IPAA is necessary. The patient then requires regular endoscopic evaluation of the rectum to remove any polyps that develop.
Although the primary focus of surveillance is the colon and rectum, some individuals with MAP may develop polyps in the upper gastrointestinal tract. Therefore, periodic upper endoscopic examinations should be performed to evaluate the esophagus, stomach, and duodenum. The frequency of upper endoscopy may vary based on individual factors and should be discussed with healthcare professionals. Baseline EGD, including visualization of the ampulla of Vater, is also recommended for patients with MAPs beginning at 30-35 years of age.
JUVENILE POLYPOSIS SYNDROME
JPS is a rare genetic disorder of BMPR1A and SMAD4, characterized by the development of multiple polyps in the gastrointestinal tract, particularly in the colon and rectum. These polyps are hamartomas that can lead to various complications including bleeding, anemia, and an increased risk of developing CRC [5,6]. Colonoscopic examinations are typically recommended for individuals with JPS, starting in early childhood or adolescence, depending on specific genetic mutations and individual circumstances. The frequency of colonoscopy ranges from 1 to 3 years. Because JPS can also involve polyp growth in the stomach and small intestine, regular EGD examinations are recommended every 1 to 3 years. The timing and frequency of upper endoscopy may vary depending on individual factors. Additional screening such as pancreatic or chest imaging may be considered in some cases.
PEUTZ-JEGHERS SYNDROME
PJS is a rare inherited syndrome with a germline mutation in STK11, which is characterized by the development of gastrointestinal polyps, characteristic mucocutaneous freckling, and an increased risk of certain cancers, including CRC, gastric pancreatic, breast, and ovarian cancer [5,6]. Intestinal polyps in the PJS are hamartomas, especially in cases of large polyps, leading to intussusception with obstruction of the small bowel.
If PJS is suspected based on clinical symptoms or family history of the syndrome, a diagnostic workup is typically performed. This may include a thorough medical history review, physical examination, endoscopy, and imaging tests, including EGD, colonoscopy, and CT scan, and genetic testing, to identify specific mutations associated with PJS. Regular surveillance of the gastrointestinal tract is crucial in individuals with PJS because of the development of hamartomatous polyps. The frequency and timing of surveillance tests may vary depending on individual factors including age, polyp burden, and other risk factors. The frequency of EGD typically ranges from 1 to 2 years, and the frequency of colonoscopy may vary, but is typically performed every 3 to 5 years. Periodic imaging tests such as capsule endoscopy or CT/magnetic resonance enterography may be considered to evaluate PJS polyps in the small intestine. The frequency of small bowel imaging may range from 1 to 2 years.
CONCLUSIONS
Screening and surveillance are essential tools for the management of hereditary CRC. By identifying individuals with a family history of CRC and implementing regular screening protocols, healthcare providers can detect precancerous and cancerous changes in the colon and rectum at early stages. Early detection allows timely intervention and treatment, which significantly improves patient outcomes and reduces mortality rates. Moreover, surveillance programs enable healthcare professionals to closely monitor disease progression and promptly address new developments. By offering comprehensive screening and surveillance strategies to at-risk individuals, healthcare systems can effectively mitigate the impact of hereditary CRC, empower patients to make informed decisions about their health, and potentially prevent the development of advanced-stage CRC.